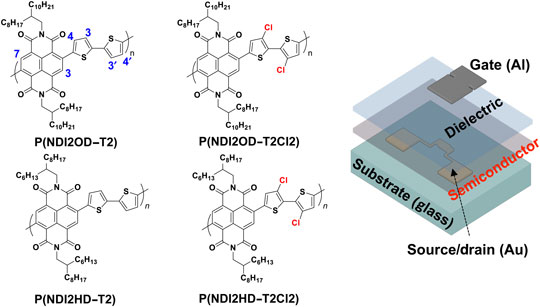
We use first-principles calculations based on density functional theory to investigate the interplay between oxygen vacancies, A-site cation size/tolerance factor, epitaxial strain, ferroelectricity, and magnetism in the perovskite manganite series, AMnO3 (A = Ca2+, Sr2+, Ba2+). We find that, as expected, increasing the volume through either chemical pressure or tensile strain generally lowers the formation energy of neutral oxygen vacancies consistent with their established tendency to expand the lattice. Increased volume also favors polar distortions, both because competing rotations of the oxygen octahedra are suppressed and because Coulomb repulsion associated with cation off-centering is reduced. Interestingly, the presence of ferroelectric polarization favors ferromagnetic (FM) over antiferromagnetic (AFM) ordering due to suppressed AFM superexchange as the polar distortion bends the Mn–O–Mn bond angles away from the optimal 180°. Intriguingly, we find that polar distortions compete with the formation of oxygen vacancies, which have a higher formation energy in the polar phases; conversely the presence of oxygen vacancies suppresses the onset of polarization. In contrast, oxygen vacancy formation energies are lower for FM than AFM orderings of the same structure type. Our findings suggest a rich and complex phase diagram, in which defect chemistry, polarization, structure, and magnetism can be modified using chemical potential, stress or pressure, and electric or magnetic fields.