
108 results
Introduction
-
-
- Book:
- Cambridge Handbook of Anesthesiology
- Published online:
- 24 May 2023
- Print publication:
- 08 June 2023, pp 1-3
-
- Chapter
- Export citation
Multi-Sun EELS: Ultra-High Energy Resolution combined with High Spatial Resolution and High Beam Current
-
- Journal:
- Microscopy and Microanalysis / Volume 28 / Issue S1 / August 2022
- Published online by Cambridge University Press:
- 22 July 2022, pp. 2640-2642
- Print publication:
- August 2022
-
- Article
-
- You have access
- Export citation
Healthcare worker safety program in a coronavirus disease 2019 (COVID-19) alternate care site: The Javits New York Medical Station experience
- Part of
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 44 / Issue 2 / February 2023
- Published online by Cambridge University Press:
- 18 April 2022, pp. 268-276
- Print publication:
- February 2023
-
- Article
- Export citation
Cosmology with Phase 1 of the Square Kilometre Array Red Book 2018: Technical specifications and performance forecasts
- Part of
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 37 / 2020
- Published online by Cambridge University Press:
- 06 March 2020, e007
-
- Article
-
- You have access
- HTML
- Export citation
-
We present a detailed overview of the cosmological surveys that we aim to carry out with Phase 1 of the Square Kilometre Array (SKA1) and the science that they will enable. We highlight three main surveys: a medium-deep continuum weak lensing and low-redshift spectroscopic HI galaxy survey over 5 000 deg2; a wide and deep continuum galaxy and HI intensity mapping (IM) survey over 20 000 deg2 from
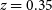 $z = 0.35$
to 3; and a deep, high-redshift HI IM survey over 100 deg2 from
$z = 0.35$
to 3; and a deep, high-redshift HI IM survey over 100 deg2 from 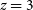 $z = 3$
to 6. Taken together, these surveys will achieve an array of important scientific goals: measuring the equation of state of dark energy out to
$z = 3$
to 6. Taken together, these surveys will achieve an array of important scientific goals: measuring the equation of state of dark energy out to 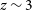 $z \sim 3$
with percent-level precision measurements of the cosmic expansion rate; constraining possible deviations from General Relativity on cosmological scales by measuring the growth rate of structure through multiple independent methods; mapping the structure of the Universe on the largest accessible scales, thus constraining fundamental properties such as isotropy, homogeneity, and non-Gaussianity; and measuring the HI density and bias out to
$z \sim 3$
with percent-level precision measurements of the cosmic expansion rate; constraining possible deviations from General Relativity on cosmological scales by measuring the growth rate of structure through multiple independent methods; mapping the structure of the Universe on the largest accessible scales, thus constraining fundamental properties such as isotropy, homogeneity, and non-Gaussianity; and measuring the HI density and bias out to 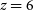 $z = 6$
. These surveys will also provide highly complementary clustering and weak lensing measurements that have independent systematic uncertainties to those of optical and near-infrared (NIR) surveys like Euclid, LSST, and WFIRST leading to a multitude of synergies that can improve constraints significantly beyond what optical or radio surveys can achieve on their own. This document, the 2018 Red Book, provides reference technical specifications, cosmological parameter forecasts, and an overview of relevant systematic effects for the three key surveys and will be regularly updated by the Cosmology Science Working Group in the run up to start of operations and the Key Science Programme of SKA1.
$z = 6$
. These surveys will also provide highly complementary clustering and weak lensing measurements that have independent systematic uncertainties to those of optical and near-infrared (NIR) surveys like Euclid, LSST, and WFIRST leading to a multitude of synergies that can improve constraints significantly beyond what optical or radio surveys can achieve on their own. This document, the 2018 Red Book, provides reference technical specifications, cosmological parameter forecasts, and an overview of relevant systematic effects for the three key surveys and will be regularly updated by the Cosmology Science Working Group in the run up to start of operations and the Key Science Programme of SKA1.
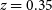
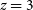
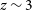
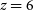
Fundamental physics with the Square Kilometre Array
- Part of
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 37 / 2020
- Published online by Cambridge University Press:
- 27 January 2020, e002
-
- Article
-
- You have access
- HTML
- Export citation
Organizational Structure and Operation of the Illinois Wine Industry
-
- Journal:
- Agricultural and Resource Economics Review / Volume 43 / Issue 1 / April 2014
- Published online by Cambridge University Press:
- 15 September 2016, pp. 104-124
-
- Article
- Export citation
Contributors
-
-
- Book:
- Tip-of-the-Tongue States and Related Phenomena
- Published online:
- 05 June 2014
- Print publication:
- 16 June 2014, pp vii-viii
-
- Chapter
- Export citation
Radio Continuum Surveys with Square Kilometre Array Pathfinders
- Part of
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 30 / 2013
- Published online by Cambridge University Press:
- 27 March 2013, e020
-
- Article
-
- You have access
- HTML
- Export citation
EMU: Evolutionary Map of the Universe
- Part of
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 28 / Issue 3 / 2011
- Published online by Cambridge University Press:
- 02 January 2013, pp. 215-248
-
- Article
-
- You have access
- Export citation
Reply to Webster and Osborne
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 32 / Issue 10 / October 2011
- Published online by Cambridge University Press:
- 02 January 2015, pp. 1047-1048
- Print publication:
- October 2011
-
- Article
-
- You have access
- Export citation
Economic Value of Dispensing Home-Based Preoperative Chlorhexidine Bathing Cloths to Prevent Surgical Site Infection
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 32 / Issue 5 / May 2011
- Published online by Cambridge University Press:
- 02 January 2015, pp. 465-471
- Print publication:
- May 2011
-
- Article
- Export citation
The Fundamental Plane of Early-Type Galaxies
-
- Journal:
- European Astronomical Society Publications Series / Volume 48 / 2011
- Published online by Cambridge University Press:
- 11 July 2011, pp. 411-412
- Print publication:
- 2011
-
- Article
- Export citation
28 - Hereditary aceruloplasminemia
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 284-289
-
- Chapter
- Export citation
Plate section
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp -
-
- Chapter
- Export citation
31 - Neuroferritinopathies
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 299-303
-
- Chapter
- Export citation
Contents
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp v-vi
-
- Chapter
- Export citation
12 - Hemochromatosis associated with ferroportin gene (SLC40A1) mutations
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 171-180
-
- Chapter
- Export citation
23 - Iron overload associated with pyruvate kinase deficiency
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 251-253
-
- Chapter
- Export citation
2 - Normal iron absorption and metabolism
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 10-27
-
- Chapter
- Export citation
20 - Divalent metal transporter-1 (SLC11A2) iron overload
-
-
- Book:
- Handbook of Iron Overload Disorders
- Published online:
- 01 June 2011
- Print publication:
- 22 July 2010, pp 228-232
-
- Chapter
- Export citation
