



A 53-year-old man with a 5-year history of untreated chronic lymphocytic leukemia (CLL) presents for assessment of neck pain, adenopathy, and urinary retention. He has known obstructive sleep apnea, gastroesophageal reflux, and had undergone chemotherapy, radiation, and bowel resection for Burkitt’s lymphoma. A magnetic resonance imaging (MRI) of his cervical spine showed an extensive, expansile, intramedullary T2-hyperintense lesion from craniocervical junction to C5–6. Postcontrast imaging of the cervical spinal cord showed extensive diffuse patchy heterogeneous enhancement along with few focal enhancing discrete nodules. A cerebrospinal fluid (CSF) analysis was consistent with CLL, and he underwent three pulse treatments with dexamethasone and one transfusion, after which he improved clinically and radiographically. Two months later, he had recurrence of his previous symptoms and also experienced sudden, profound, monocular vision loss in his right eye. An MRI of his orbits showed T2 hyperintensity of the right optic nerve, within the orbit extending toward the orbital apex as well as within the intraconal fat surrounding the optic nerve. An MRI of his brain was otherwise normal. Examination revealed a relative afferent pupillary defect with visual acuity of 20/200 on the right and 20/15 on the left. He was diffusely hyper-reflexic, his plantar responses were upgoing bilaterally, and he had a spastic gait. His cranial nerve, motor, sensory, and coordination examinations were normal. Digital rectal examination revealed normal rectal tone and sensation, and a post-void residual at the time showed retention of 500 mL. Repeat CSF analysis revealed normal glucose, elevated protein (2.09 g/L), elevated total nucleated cells (73 × 106) and slightly elevated erythrocytes (70 × 106/L). Flow cytology showed a monotonous population of small lymphocytes, and cytometry showed a monoclonal B-cell population expressing cytoplasmic kappa light chains, consistent with CLL. A repeat MRI of his spine showed diffuse, abnormal / T2 hyperintensity throughout the central cord extending from the medulla to the conus (Figure 1). Autoimmune workup included antinuclear antibody (ANA) screen, complement levels, and immunoglobulins (Igs), rheumatoid factor, a paraneoplastic panel (anti-Hu, anti-Ri, and anti-Yo) and HLA typing. The IgG was elevated at 26 g/L (normal 6.35–14.65 g/L), and both IgA and IgM were low but considered to be within error for the lab values. The ANA screen included DNA, ANCAs, chromatin, ribo protein, SS-A52, SS-A60, SS-B, centromere-B, Sm, SmRNP, RNP68, RNP A, SCL-70, and JO-1. The ANA screen for the RNP A being 1.1 (normal <1.0) was positive. Infectious workup included CSF Gram stain and culture, enterovirus PCR, HSV 1 and 2 PCR, and VZV PCR. Blood was sent for analyzing HTLV 1 and 2 antibodies and CMV IgG antibodies but were reported as negative for all. Interestingly, serum anti-aquaporin-4 (AQP4) antibodies were also negative, which was confirmed using the cell-based indirect immunofluorescence testing method. Anti-MOG, amphiphysin, and anti-CRMP5 were not tested at the time. He showed minimal improvement with dexamethasone, so was started on rituximab and cyclophosphamide for treatment of his underlying CLL. He experienced complete resolution of all symptoms and remained asymptomatic at 6 months of follow-up.
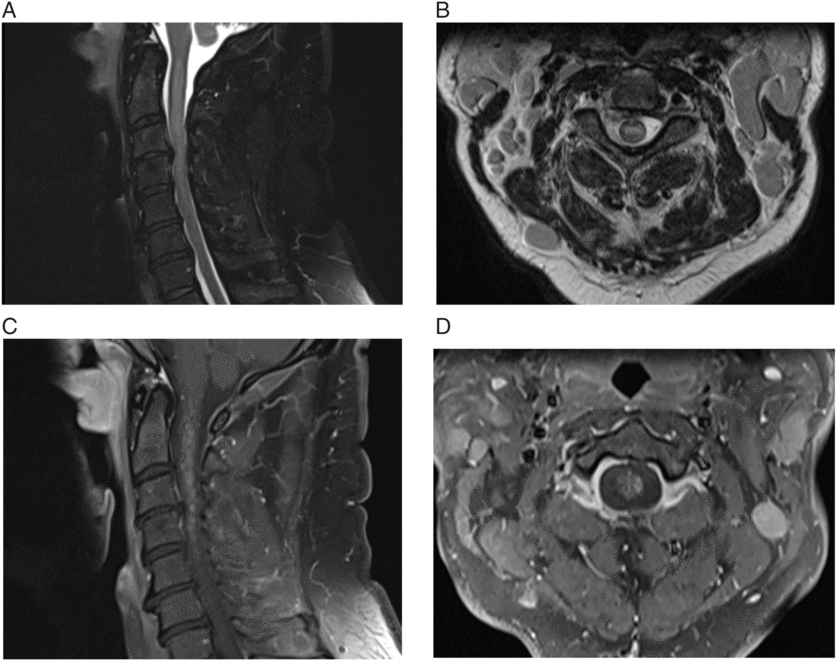
Figure 1: (A & B) Sagittal and axial T2-weighted images showing expansile intramedullary hyperintensity. (C & D) Sagittal and axial T1-weighted image, postgadolinium, showing extensive patchy diffuse heterogeneous enhancement along with focal nodular enhancement.
CLL is an abnormal proliferation of monoclonal B-type lymphocytes and is the most common form of adult-onset leukemia. Neurological complications of CLL are rare and include peripheral neuropathy, progressive multifocal leukoencephalopathy, encephalitis, neuromuscular junction dysfunction, and paraneoplastic conditions. Autoimmune complications of CLL are thought to be due to disruption in the central immune system and aberrant antigen recognition.Reference Maverakis, Goodarzi, Wehrli, Ono and Garcia 1 Manifestations are most commonly hematologic conditionsReference Hamblin 2 including autoimmune hemolytic anemia and immune thrombocytopenia.Reference Annus, Bencski and Obál 3
Neuromyelitis optica (NMO) is an autoimmune-mediated, inflammatory, demyelinating disease of the central nervous system (CNS) typically characterized by recurrent attacks of optic neuritis and a longitudinally extensive, demyelinating, spinal cord lesion.Reference Wingerchuk, Hogancamp, O’Brien and Weinshenker 4 It is caused by autoantibodies directed against the aquaporin 4 membrane protein.Reference Graber, Levy, Kerr and Wade 5 Various infections and auto-immune conditions have been associated with the subsequent development of NMO spectrum disorders (NMOSDs), and it has been suggested that such patients may have a genetic predisposition to these conditions due to the inherently abnormal autoimmunity.Reference Mandler 6 , Reference Iyer, Elsone, Appleton and Jacob 7 Although our patient meets diagnostic criteria for seronegative NMO as outlined by the international consensus,Reference Wingerchuk, Banwell and Bennett 8 there were some atypical clinical and paraclinical findings. A paraneoplastic NMO phenomenon has been described previously in association with some carcinomas, monoclonal gammopathy, some endocrinological tumors, and acute myeloid leukemia, though not with CLL.Reference Cai, He, Chu, Dai, Xu and Zhang 9
Certain features can help distinguish direct CNS involvement of CLL versus paraneoplastic etiology. In our case, the monotonous monoclonal B cells detected in the CSF implicated the underlying CLL as a cause. The pattern of enhancement can also give a clue; NMO can have patchy enhancement,Reference Krampla, Aboul-Enein and Jecel 10 but nodular enhancement within the spinal cord is indicative of cancer.Reference Loughrey, Collins, Todd, Brown and Johnson 11 In contrast, paraneoplastic myelopathy usually shows symmetric enhancement.Reference Flanagan, McKeon and Lennon 12 Our patient improved to baseline after undergoing treatment of his CLL, further suggesting either an autoimmune or a neoplastic etiology of his condition.
Our case is unique in that there was simultaneous longitudinally extensive spinal cord as well as optic nerve involvement. To our knowledge, neither a seronegative NMO-like presentation nor an NMOSD-like condition has been described previously as a manifestation of CLL.
Author Contributions
LAW, case study concept, patient examination and interpretation of data, literature review, manuscript drafting. SM, patient examination and interpretation of data, critical revision for intellectual content. KSP, patient examination and interpretation of data, critical revision for intellectual content.
Author Disclosures
None of the authors have any disclosures to report.

