



Writer’s cramp is an involuntary, sustained posture or contraction of the hand, fingers, or arm muscles while writing, typically occurring in adulthood. Reference Albanese, Bhatia and Bressman1 Childhood-onset has mainly been described as a feature of myoclonus-dystonia syndrome, Reference Gerrits, Foncke, Koelman and Tijssen2 besides isolated cases of dopa-responsive dystonia. Reference Deonna, Roulet, Ghika and Zesiger3 Hereditary spastic paraplegia type 15 (SPG15) (OMIM#270700) is a complex, autosomal recessive form of spastic paraplegia caused by pathogenic variants in ZFYVE26 (OMIM * 612012). Reference Hanein, Martin and Boukhris4,Reference Denora, Muglia and Casali5 Here, we intend to describe childhood-onset writer’s cramp as part of SPG15 phenotype.
We describe a 7-year-old righthanded boy, from consanguineous parents, with hands tremor first noticed when he entered elementary school. This initially affected his left hand but rapidly progressed to the right one; was mainly elicited by writing and accompanied by abnormal posturing. He had normal perinatal history and psychomotor development and no known family history of neurological disorders. During his first year of schooling, learning problems emerged, leading to premature drop-out. At age 13 his speech became slurred, and there was progressive gait impairment, with frequent falls. Two years later, he had a left predominant postural and kinetic upper limb (UL) tremor, with bilateral dystonic posturing when writing (video – segment 1), and generalized hyperreflexia. A trial of levodopa showed no improvement. At age 16 years, his 10-year-old sister developed spastic paraplegia (Figure 1 – family pedigree). Over the years, he presented progressive cognitive and motor deterioration, becoming wheelchair-bound by his mid-twenties. At the last visit (age 32 years), neurological examination showed dementia, spastic dysarthria, distal atrophies, spastic tetraparesis with hyperreflexia, bilateral Babinski sign and truncal anteflexion (video – segment 2). An extensive investigation was conducted over time. Brain MRI (ages 15 and 19) revealed a thin corpus callosum. At age 15, a metabolic panel (serum very-long-chain fatty acids, pyruvate, lactate, copper, ceruloplasmin; CSF neurotransmitters) was unremarkable; and no pathogenic variants were identified in TOR1A. Later, a study of ZFYVE26 by Sanger sequencing showed a novel, likely-pathogenic (American College of Medical Genetics and Genomics classification criteria PVS1 and PM2 are applicable) variant on exon 25 (NM_015346.4:c.4914_4917del(p. Phe1638Leufs*4)) in homozygosity. This genotype was also present in his sister, their parents being heterozygotes.
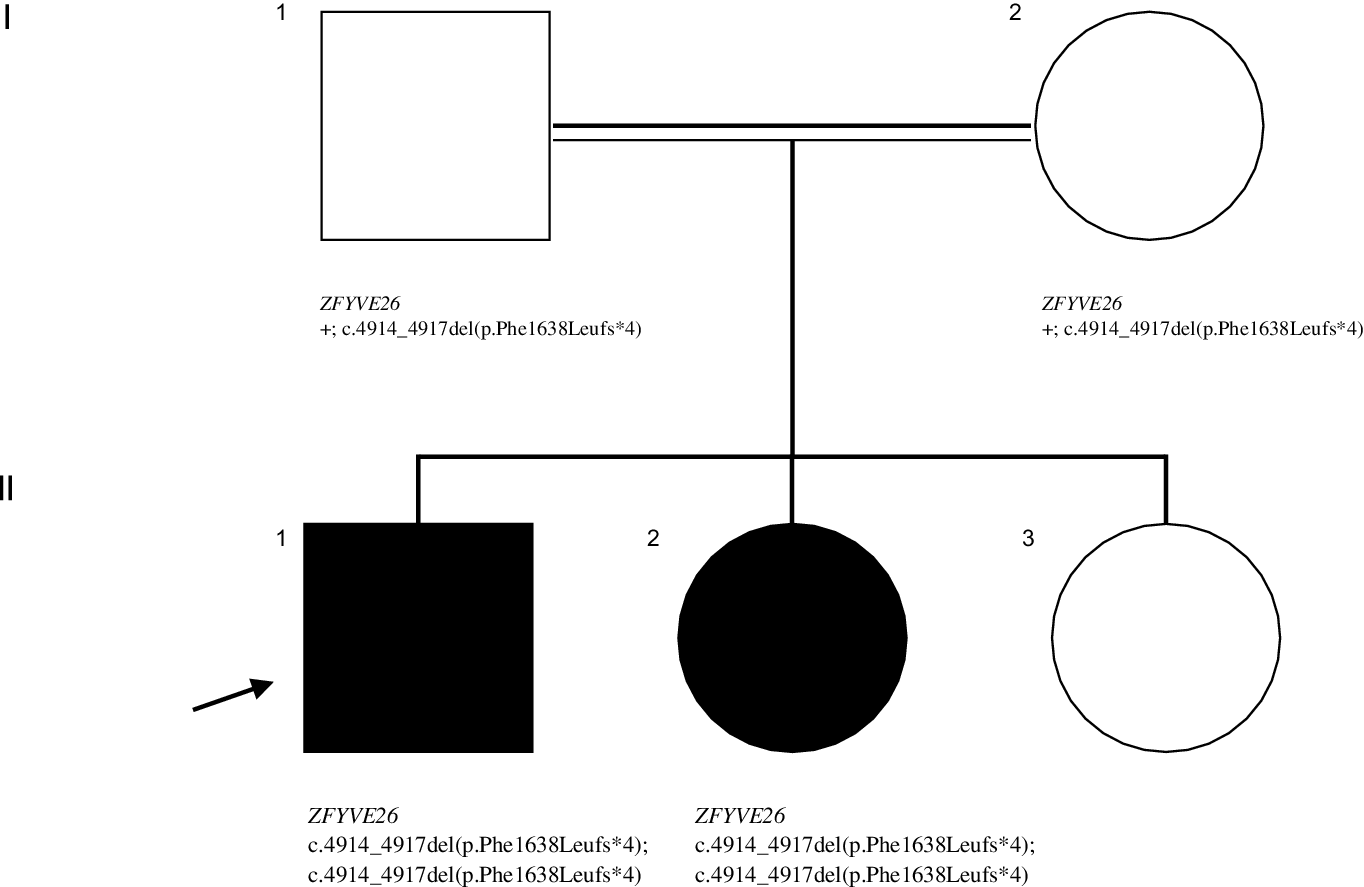
Figure 1: Pedigree showing the segregation of the ZFYVE2 variant. Symbols: arrow – proband; filled symbol- affected individual; open symbol- unaffected individual; + - wild type allele.
This case concerns a young patient presenting with UL tremor and writer’s cramp, latter developing cognitive impairment and pyramidal signs, whose investigation led to the identification of a likely-pathogenic variant on ZFYVE26. In addition to bilateral lower limb spasticity and hyperreflexia, SPG15 usually encompasses cognitive deterioration preceding pyramidal signs, Reference Hanein, Martin and Boukhris4 as occurred in this patient. Mean age of onset is usually below 15 years, and seizures, movement disorders, or peripheral neuropathy may be present in a considerable number of patients. Reference Hanein, Martin and Boukhris4 To the best of our knowledge, limb dystonia has been only described in another family of four affected individuals, with one presenting hand dystonia without a task-specific nature. Reference Hanein, Martin and Boukhris4 It is very interesting that in both our family and the previously reported, dystonia was present in only one member. Reference Hanein, Martin and Boukhris4 Such intra-familial variability has been recognized in several neurological disorders and raises the role of epigenetic factors, yet unidentified, in its clinical expression.
Before the widespread use of next-generation sequencing (NGS), dystonia has very occasionally been described in patients with recessive, complex SPG forms. Coutinho et al. (2009) identified only one case of dystonia (and a complex phenotype) in a series of 106 patients with autosomal recessive spastic paraplegia. Reference Coutinho, Barros and Zemmouri6 More recently, spasmodic dysphonia, cervical and limb dystonia were reported in SPG7, and oromandibular dystonia in SPG11. Reference Schaefer, Szekely, Moeller and Tinaz7 In the era of NGS, dystonia is being increasingly recognized in complex forms, as SPG76 or SPG78, either preceding pyramidal signs or emerging later in disease course. Reference Odake, Koh and Takiyama8 Still, a task-specific nature at presentation has not been reported thus far.
Spastizin, the protein encoded by ZFYVE26, appears to play a role in endosomal trafficking, given its colocalization with markers of the endoplasmic reticulum and endosomes. Reference Hanein, Martin and Boukhris4 Loss of function of spastizin (similarly to spatacsin) has also been implicated in the accumulation of autolysosomes and depletion of free lysosomes, translating a failure of autophagic lysosome reformation. Reference Chang, Lee and Blackstone9 This may provide the background for the underlying pathogenic mechanism of dystonia in SPG15, as ultrastructural lysosomal abnormalities have been implicated in early-onset dystonia. We believe it would be interesting to perform functional studies on endosomal trafficking and lysosomal function in HSP patients with and without dystonia.
Herein, we describe a unique presentation of a patient with a ZFYVE26 homozygous variant, with records spanning 17 years of disease, documenting the progression of a childhood-onset focal, task-specific dystonia, to a typical phenotype of spastic paraplegia with thin corpus callosum, with predominant cognitive and pyramidal signs. This report broadens the phenotypic spectrum of ZFYVE26-related disorders and highlights the increasingly recognized insufficiency of phenotype-based classification systems for hereditary neurological diseases.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2022.59.
Conflicts of Interest
Joana Damásio acted as consultant for Bial and Zambon pharmaceuticals. The remaining authors have no competing interests to report.
Statement of Authorship
JM – Conception, organization and execution of the study; Writing of the first draft.
AS - Conception, organization and execution of the study.
JO – Acquisition and analysis of data; Review and critique of the manuscript.
AM - Acquisition and analysis of data; Review and critique of the manuscript.
JB - Acquisition and analysis of data; Review and critique of the manuscript.
JS - Acquisition and analysis of data; Review and critique of the manuscript.
CB - Acquisition and analysis of data; Review and critique of the manuscript.
JD - Conception, organization and execution of the study; Review and critique of the manuscript.
Availability of Data and Material
Research data have not been archived at a public repository, but are available upon request at CHUPorto.
Ethics Approval
This project has been approved by the ethical committee of CHUPorto. Patient’s informed consent was obtained, according to the declaration of Helsinki, for video recording and publication.
Consent to Participate
The patient’s parents provided written informed consent to participate in the study.
Consent for Publication
The patient’s parents provided written informed consent for publication and video recording and publication.



