


Antioxidants are substances that counteract free radicals and prevent the damage caused by them. In recent years, antioxidants have emerged as promising prophylactic and therapeutic agents(Reference Ratnam, Ankola and Bhardwaj1). Nicotinic acid (NA), also known as niacin (vitamin B3), is considered as a major antioxidant since it influences multiple pathways tied to both cellular survival and cellular death. During oxidative stress (OS), NA protects the cell by blocking cellular inflammatory cell activation, early apoptotic phosphatidylserine exposure and late nuclear DNA degradation(Reference Maiese, Chong and Hou2). NA supplementation has been shown to reverse OS-induced cell injury in kidney epithelial HEK 293 cells(Reference Hara, Yamada and Shibata3). Zn2+ is another major antioxidant that offers protection through the antagonism of redox-active transition metals, such as Cu and Fe, the induction of metallothionein synthesis, the guarding of protein sulfhydryl groups from oxidative damage(Reference Prasad, Bao and Beck4, Reference Powell5) and the stabilisation of Cu–Zn superoxide dismutase (SOD)(Reference Andrews6). Zn is necessary for normal liver function, as Zn deficiency could participate in the pathogenesis of liver diseases(Reference Stamoulis, Kouraklis and Theocharis7) and reduced hepatic Zn levels have been correlated with impaired liver function and regeneration(Reference Grungreiff8). It has also been reported that Zn deficiency leads to a rapid increase in cellular oxidants(Reference Oteiza, Clegg and Zago9, Reference Ho and Ames10).
Vitamins such as riboflavin, NA, thiamin, folic acid and ascorbic acid have functional groups reported to be capable of forming complexes with Zn. Our previous studies with erythrocytes(Reference Agte, Nagmote and Chiplonkar11), Caco-2 cells(Reference Tupe and Agte12) and hepatocytes(Reference Tupe, Chiplonkar and Agte13) have demonstrated the effects of NA, folic acid and ascorbic acid on Zn bioavailability, indicating interactions between vitamins and Zn. The results from in vitro experiments demonstrated that improved Zn metabolism can be achieved through enhanced Zn absorption (intestinal) and post-absorptive uptake (hepatic) especially under OS conditions in the presence of NA. Another study from our laboratory has revealed that NA supplementation as a finger millet-based diet resulted in a significant enhancement of Zn absorption, hepatic Zn levels and growth of weanling mice(Reference Agte, Paknikar and Chiplonkar14). In a recent study, we have reported a dose-dependent increase in hepatic NA accumulation as dietary Zn levels increased from deficient to excess(Reference Tupe, Tupe and Tarwadi15). The interaction between Zn and NA seems to be bidirectional. Therefore, it was thought worthwhile to explore the efficacy of NA administration in attenuating the adverse effects caused by OS exposure to a Zn-deficient state in vivo. Hence, animal experiments were conducted wherein the effect of NA on Zn metabolism under Zn deficiency and OS in growing rats was investigated, and the findings are reported in the present study.
Experimental methods
Animals, diet and experimental design
Weanling male Wistar rats weighing 50 (sd 10) g were procured from the animal facility, Agharkar Research Institute, Pune. The animal experiments' protocol was approved by the Agharkar Research Institute's Institutional Animal Ethics Committee, and the rats were treated according to the guidelines set by the Committee for the Purpose of Control and Supervision of Experiments on Animals.
The animals were housed individually in polypropylene cages in the institute's animal house under hygienic conditions in a room maintained at 24 ± 2°C and with a 12 h light–12 h dark cycle. During the study period of 3 weeks, the rats were fed with a modified AIN-93G diet prepared according to the American Institute of Nutrition guidelines(Reference Reeves, Nielsen and Fahey16), containing casein as the source of protein, wheat bran as the source of fibre, and maize starch and sucrose as the sources of carbohydrates. All rats were fed the treatment diets and distilled water ad libitum, throughout the experimental period. A weekly record of body-weight changes and food intake of rats for all groups was maintained. The only variations were that Zn content in the diet (added as ZnSO4) was at a marginally deficient level (10 mg Zn/kg diet), and NA was given at three levels, namely deficient (10 mg NA/kg diet), adequate (30 mg NA/kg diet) and excess (1000 mg NA/kg diet). The reported excess dietary NA supplementations were from 500 and 1000 mg/kg diet(Reference Jackson, Rawling and Roebuck17) to pharmacologically supplemented 4 g NA/kg diet(Reference Boyonoski, Spronck and Jacobs18).
The study design and grouping of animals for the experiment are shown in Fig. 1. Group I (GI, n 8) animals served as normal controls and were fed with an AIN-93G diet containing adequate Zn and NA (30 mg each/kg diet). The remaining animals were randomly divided into two main groups, groups II and III (GII and GIII), which were with OS and without OS, respectively. NA supplementation may improve Zn metabolism and prevent or cure OS-induced damage, hence it was decided to assess this by inducing OS before and after the dietary NA addition, respectively. Considering these variables, the main group GII was subdivided into two sets depending on the OS treatment (intraperitoneal (i.p.) injections of tert-butyl hydroperoxide) within the experimental period of 3 weeks, which were (1) pre-exposure (OS treatment in the first week only) and (2) post-exposure (OS treatment in the third week). At each OS exposure condition, dietary NA levels varied as deficient, normal and excess states, which resulted in a total of six treatment subgroups (n 8), i.e. GIIa, GIIb and GIIc for pre-exposure and GIId, GIIe and GIIf for post-exposure. The corresponding six control groups (GIIIa–GIIIf) were also maintained, wherein only NA variation was done without any OS treatment (i.p. injections of saline). For pre-exposure, GIIa, GIIb and GIIc animals were given OS treatment by i.p. injections of 0·22 mmol/kg body weight of tert-butyl hydroperoxide on the 2nd, 4th and 6th day of the first week. The respective controls GIIIa, GIIIb and GIIIc were given i.p. injections of saline on the same days. All these groups were given a normal American Institute of Nutrition diet (containing 30 mg Zn and NA each/kg diet) during the first week, whereas for the next 2 weeks, the groups were maintained on a specific diet assigned to each group (marginally deficient Zn with respective NA levels – deficient, normal and excess). For post-exposure, animals were maintained on a specified diet for the first 2 weeks, and a normal diet and the treatment of three i.p. injections (one every alternate day) of tert-butyl hydroperoxide (for GIId, GIIe and GIIf) or saline (for GIIId, GIIIe and GIIIf) were given in the third week. The diet and OS treatment schedule for GII and GIII animals is shown in Fig. 2. The design was thought to help in better understanding the role of NA supplementation in Zn metabolism and OS.

Fig. 1 Design of an in vivo animal experiment for studying the effect of nicotinic acid (NA) supplementation on zinc metabolism under different oxidative stress (OS) exposure conditions. NA D, NA deficient; NA N, NA normal; NA E, NA excess; GI, group I; GII, group II; GIII, group III.

Fig. 2 Oxidative stress (OS) and diet schedule for groups II and III animals during the (a) pre-exposure and (b) post-exposure OS treatment. i.p., Intraperitoneal; t-BHP, tert-butyl hydroperoxide; NA D, nicotinic acid deficient; NA N, NA normal; NA E, NA excess; ZnD, zinc deficient.
Tissue sampling
After 3 weeks, the rats were deprived of food for 6 h and then euthanised under light diethyl ether anaesthesia. Blood was collected by cardiac puncture into EDTA-containing tubes. After collecting the blood, the animals were dissected, and the livers were excised and rinsed in PBS. A small section of each liver was placed in 10 % phosphate-buffered formalin for histopathological analysis, and the remaining liver tissue was processed for various biochemical estimations. The blood samples were centrifuged at 4000 rpm for 10 min. The plasma was separated and processed on the same day for the estimation of liver marker enzymes, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT).
Estimation of liver marker enzymes in plasma
The enzyme activities of AST and ALT were estimated using commercial kits (Ranbaxy Diagnostics Ltd, Baddi, H.P., India) as described previously(Reference Tupe, Tupe and Tarwadi15), and the enzyme activities are expressed in terms of IU/l of plasma at 37°C.
Histopathological studies
For histopathology, liver samples fixed in formalin were dehydrated in ascending grades of alcohol, cleared in benzene and embedded in paraffin wax. The blocks were cut into 5–7 μm thin sections, which were then double-stained with haematoxylin and eosin. After staining, the sections were observed under a light microscope and photographed.
Biochemical analysis
The liver samples were homogenised in 100 mm-potassium phosphate buffer (pH 7·5) containing 0·15 m-KCl with a Potter–Elvehjem homogeniser to obtain 10 % homogenate. Tissue homogenates were centrifuged at 10 000 g for 30 min at 4°C, and the supernatants were used for the estimation of different enzymes, protein, reduced glutathione (GSH) and lipid peroxidation.
Catalase, SOD and glutathione peroxidase (GPx) activities were determined by following the methods of Clairborne & Fridovich(Reference Clairborne and Fridovich19), Kono(Reference Kono20) and Mohondas et al. (Reference Mohandas, Marshell and Duggin21), respectively. Protein concentrations were determined by the method of Lowry et al. (Reference Lowry, Rosebrough and Farr22), and the levels of reduced GSH were estimated by using the method of Ellman(Reference Ellman23). The estimation of lipid peroxidation in the liver was done by the method of Placer et al. (Reference Placer, Crushman and Johnson24). All these methods have been described briefly in our previous report(Reference Tupe, Tupe and Tarwadi15).
Hepatic zinc estimation
Zn concentrations in liver samples were measured as described earlier(Reference Tupe, Tupe and Tarwadi15) using atomic absorption spectrophotometry (AA 800; Perkin-Elmer, Shelton, CT, USA).
Data presentation and statistical analysis
Data are presented as means and standard deviations. The experimental parameters from the OS treatment group (GII) and without OS exposure groups (GIII) were initially compared with the normal control (GI) using one-way ANOVA. The effect of NA supplementation and OS exposure in the treatment groups (GII and GIII) was then assessed using two-way ANOVA. Student's unpaired t test was used to further analyse differences between group pairs (i.e. NA-supplemented groups and OS exposure groups). To compare the effect of OS treatment, the subgroups from GII were compared with the respective subgroups from GIII, for example GIIa with GIIIa. For comparison of NA supplementation within the GII and GIII groups, the NA-deficient groups (GIIa and GIIIa) were compared with the respective NA normal and NA excess groups.
Results
Body weight and food intake records for the OS pre-exposure experiment showed no significant difference in the different treatment groups and their subgroups. GII animals with OS treatment showed a slight decrease (NS) in weights and intake, indicating that OS pre-exposure along with Zn deficiency and dietary NA variation was tolerated by the animals.
Effect on aspartate aminotransferase and alanine aminotransferase
The consequences of NA variation in the diet under Zn deficiency and OS pre-exposure on plasma AST and ALT levels are shown in Fig. 3. OS pre-exposure to the rats caused significant elevations in plasma ALT and AST levels in GII animals compared with the normal control group (GI). The highest AST and ALT levels were observed in GIIa (P < 0·001 v. GI) animals, indicating increased hepatotoxicity due to tert-butyl hydroperoxide coupled with Zn and NA deficiency. Enzyme activities were reduced (P < 0·01) in GIIb and GIIc animals as the dietary NA level changed from the deficient to normal to excess states, respectively. Compared with GI animals, these enzymes were slightly elevated in GIIIa (P < 0·05) animals, indicating that even without OS treatment, the Zn-deficient diet affects the levels of liver marker enzymes. Moreover, in GIIIc animals, the Zn-deficient but NA excess diet showed comparable activities with GI animals.
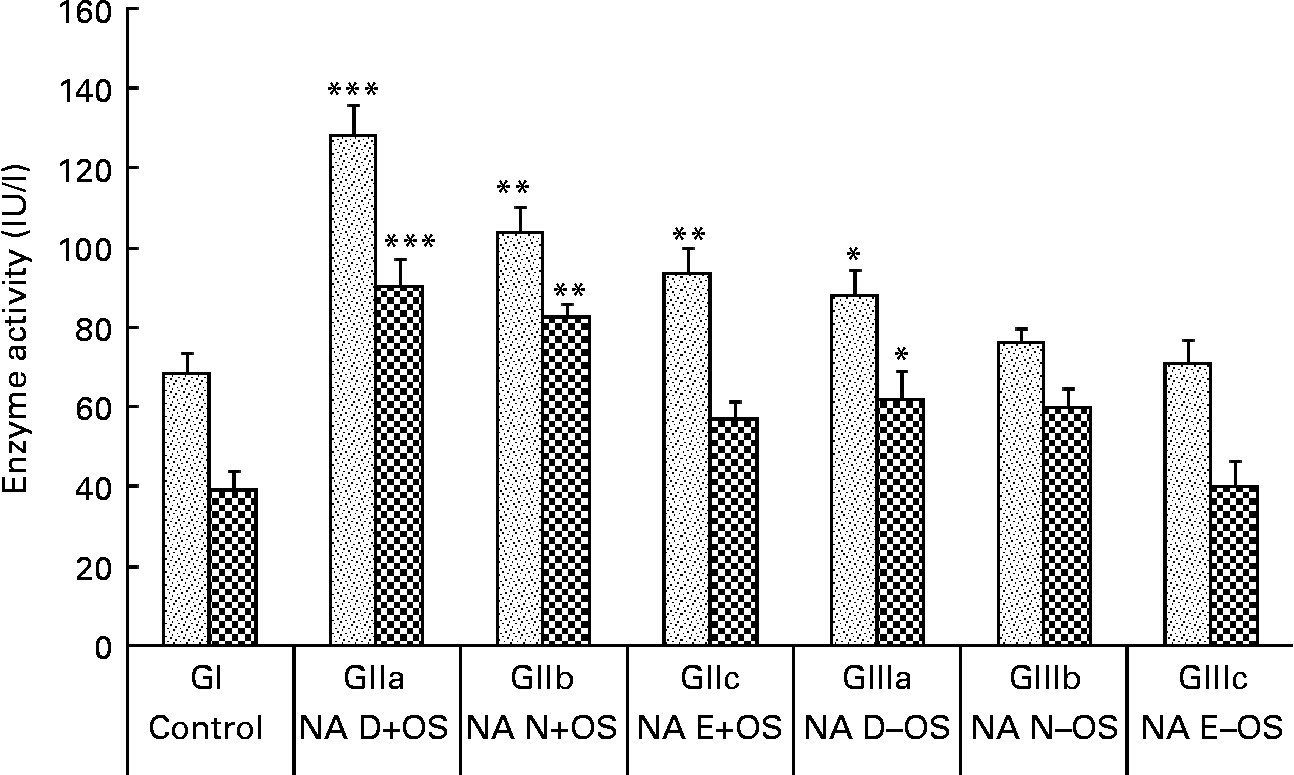
Fig. 3 Effect of dietary nicotinic acid (NA) variation on plasma enzymes aspartate aminotransferase (![]() ) and alanine aminotransferase (
) and alanine aminotransferase (![]() ) in animals treated with pre-exposure to oxidative stress (OS) conditions. Values are means, with standard deviations represented by vertical bars. NA D, NA deficient; NA N, NA normal; NA E, NA excess; GI, group I; GII, group II; GIII, group III. Mean value was significantly different from that of the normal control group (GI): *P<0·05, **P<0·01, ***P<0·001.
) in animals treated with pre-exposure to oxidative stress (OS) conditions. Values are means, with standard deviations represented by vertical bars. NA D, NA deficient; NA N, NA normal; NA E, NA excess; GI, group I; GII, group II; GIII, group III. Mean value was significantly different from that of the normal control group (GI): *P<0·05, **P<0·01, ***P<0·001.
OS post-exposure to the rats resulted in elevated levels of AST (145·39 (sd 8·1) IU/l) and ALT (113·8101 (sd 5·7) IU/l) in GIId animals, which remained towards the higher side even after excess NA supplementation in GIIf (AST: 116·4 (sd 2·5) IU/l; ALT: 71·295 (sd 3·9) IU/l) animals.
Histopathological observations
The effect of different treatments, i.e. OS pre- and post-exposure with dietary NA variations, on liver morphology of selected groups is shown in Fig. 4. Liver from control rats (GI) showed preserved hepatic architecture with normal hepatocytes (Fig. 4(e)). In the rat liver from OS pre-exposure with the NA-deficiency group, it was observed that the hepatocytes showed necrosis with cytoplasmic vacuolisation. A dilated central vein was also observed in this sample (Fig. 4(a)). However, these effects were almost diminished by excess NA supplementation in OS pre-exposure, since nearly preserved hepatic architecture with normal hepatocytes was observed (Fig. 4(b)). Maximum tissue destruction was evident in OS post-exposure with NA deficiency (Fig. 4(c)). Peritoneal fibrosis with a mononuclear cell inflammatory infiltrate was observed in the liver sample. With enhanced NA supplementation, tissue morphology was somewhat recovered, as a few focal areas of the vacuolar degeneration of hepatocytes with some dilated blood vessels were manifested (Fig. 4(d)).

Fig. 4 Effect of oxidative stress (OS) treatments with dietary nicotinic acid (NA) variation on hepatic histoarchitecture in (a) OS pre-exposure with the NA deficient (D)+Zn D group; (b) OS pre-exposure with the NA excess (E)+Zn D group; (c) OS post-exposure with the NA D+Zn D group; (d) OS post-exposure with the NA E+Zn D group; (e) control group Zn N+NA N (original magnification, 100 × ; scale bar, 50 μm).
Effect of dietary nicotinic acid levels on the hepatic antioxidant enzymes, glutathione, lipid peroxidation and zinc content
The results of OS pre- and post-exposure along with dietary NA levels on the hepatic primary antioxidant enzymes, lipid peroxidation, Zn and GSH levels are summarised in Tables 1 and 2, respectively.
Table 1 Effect of dietary nicotinic acid (NA) variations on the hepatic antioxidant enzymes, glutathione (GSH), lipid peroxidation and zinc content in rats subjected to oxidative stress (OS) pre-exposure
(Mean values and standard deviations, n 8)

SOD, superoxide dismutase; GPx, glutathione peroxidase; MDA, malondialdehyde; GI, group I; GII, group II; GIII, group III; D, deficient; N, normal; E, excess.
Mean values of all subgroups were significantly different from those of normal control GI: *P < 0·05, **P < 0·01, ***P < 0·001.
Mean values of GIIa–GIIc subgroups were significantly different from those of the GIIIa–GIIIc subgroups (for OS treatment): †P < 0·05, ††P < 0·01, †††P < 0·001.
Mean values of the GIIb–GIIc and GIIIb–GIIIc subgroups were significantly different from those of the GIIa and GIIIa subgroups (for NA variation): ‡P < 0·05, ‡‡P < 0·01, ‡‡‡P < 0·01.
§ P<0·05.
∥ P<0·001.
¶ One unit (U) of enzyme is inverse of the amount of protein required to inhibit the reduction rate of nitroblue tetrazolium by 50 %.
¶¶ Results of two-way ANOVA, as indicated by the F value.
Table 2 Effect of dietary nicotinic acid (NA) variations on the hepatic antioxidant enzymes, gluthathione (GSH), lipid peroxidation and zinc content in rats subjected to oxidative stress (OS) post-exposure
(Mean values and standard deviations, n 8)

SOD, superoxide dismutase; GPx, glutathione peroxidase; MDA, malondialdehyde; GI, group I; GII, group II; GIII, group III; D, deficient; N, normal; E, excess.
Mean values of all subgroups were significantly different from those of normal control GI: *P < 0·05, **P < 0·01, ***P < 0·001.
Mean values of GIId–GIIf subgroups were significantly different from those of the GIIId–GIIIf subgroups (for OS treatment): †P < 0·05, ††P < 0·01, †††P < 0·001.
Mean values of the GIIe and GIIf subgroups were significantly different from those of the GIId subgroup (for NA variation): ‡P < 0·05, ‡‡P < 0·01, ‡‡‡P < 0·01.
§ P<0·001.
∥ P<0·01.
¶ One unit (U) of enzyme is inverse of the amount of protein required to inhibit the reduction rate of nitroblue tetrazolium by 50 %.
¶¶ Results of two-way ANOVA, as indicated by the F value.
Comparison of group II and group III with normal control group I for the oxidative stress pre-exposure subgroups
OS pre-exposure to GIIa (with Zn-deficient and NA-deficient) animals resulted in significant (P < 0·001) depletion in SOD, catalase, GPx, GSH, Zn levels and elevated lipid peroxidation compared with normal control GI. The effect was less pronounced in GIIb and GIIc animals with normal and excess NA supplementation. Without OS treatment, the GIIIb and GIIIc groups showed a non-significant difference in the levels of all parameters compared with the GI group. However, the Zn and NA deficiency combination without OS in the GIIIa subgroup caused a decrease in GSH (P < 0·001), Zn (P < 0·01), SOD (P < 0·05) and GPx (P < 0·05) and an increase in lipid peroxidation (P < 0·05) levels.
Comparison of oxidative stress treatment between group II and group III animals for the oxidative stress pre-exposure subgroups
OS pre-exposure along with Zn and NA deficiency further worsened the antioxidant status in GIIa as against GIIIa animals. GIIa animals had decreased SOD, catalase (P < 0·001), GSH (P < 0·01), GPx and Zn (P < 0·05) levels and increased lipid peroxidation (P < 0·001) level compared with GIIIa animals. The OS treatment in GIIb and GIIc animals also caused a marginal decrease in the levels of enzymes, Zn and raised lipid peroxidation compared with GIIIb and GIIIc animals, respectively.
Intra-group comparison of nicotinc acid variation for the oxidative stress pre-exposure subgroups
As dietary NA was varied from deficient to normal to excess in GIIa, GIIb and GIIc animals, respectively, the levels of all parameters restored towards normal control. A similar trend was observed in GIII animals; however, the effect was more prominent under OS treatment, i.e. in GII animals than without OS treatment in GIII animals (Fig. 5).

Fig. 5 Effect of dietary nicotinic acid (NA) variations (NA deficient (D), NA normal (N) and NA excess (E)) on the hepatic antioxidant enzymes ((a) superoxide dismutase (SOD), (b) catalase and (c) glutathione peroxidase (GPx)), (d) lipid peroxidation, (e) glutathione and (f) Zn contents in rats subjected to treatments. (a) OS pre-exposure groups GIIa, GIIb and GIIc (![]() ); (b) OS post-exposure groups GIId, GIIe and GIIf (
); (b) OS post-exposure groups GIId, GIIe and GIIf (![]() ); (c) without OS pre-exposure groups GIIIa, GIIIb and GIIIc (
); (c) without OS pre-exposure groups GIIIa, GIIIb and GIIIc (![]() ); (d) without OS post-exposure groups GIIId, GIIIe and GIIIf (
); (d) without OS post-exposure groups GIIId, GIIIe and GIIIf (![]() ). GI, group I; GII, group II; GIII, group III.
). GI, group I; GII, group II; GIII, group III.
OS post-exposure also resulted in depleted enzymes, Zn levels and elevated lipid peroxidation for GIId animals. Recovery from this OS insult was substantially reduced after normal or excess NA supplementation (in GIIe and GIIf animals) as evident from the significantly different values than control GI. Similar to pre-exposure, NA supplementation without OS treatment across GIII in post-exposure proved beneficial as the levels of the parameters for GIIIf were almost similar to GI.
In comparison with normal control GI, the levels of SOD, catalase, GPx, glutathione and Zn were lower, whereas lipid peroxidation values were higher for all GIII subgroups. The reason may be the Zn-deficient diet given to these groups. Second, the antioxidant status of GIII from the pre-exposure experiment was better than that from the post-exposure experiment.
Discussion
The increased AST and ALT levels in GIIa and GIId animals with OS treatment and Zn deficiency indicated significant liver damage, which is in agreement with earlier reports(Reference Parsons and Disilvestro25, Reference Ishikawa, Kudo and Suzuki26). The restoration of AST and ALT levels due to normal and excess NA levels suggests its possible role in the recovery of normal liver function under Zn deficiency and OS. This may be directly due to improved Zn uptake as observed in an in vitro study(Reference Agte, Nagmote and Chiplonkar11). Liver histopathological observations, where the least damage was observed in rats with OS pre-exposure and fed with an excess NA diet afterwards, also support the beneficial role of NA in OS pre-exposure condition. However, NA supplementation before OS post-exposure did not ensure the total prevention of oxidative damage.
The present study indicated a decrease in the activity of SOD in GIIa and GIIIa animals. Several studies have shown that Zn deficiency leads to a significant decrease in SOD activity in various tissues(Reference Cao and Chen27–Reference Olin, Golup and Gershwin29). However, NA supplementation in GIIc animals significantly improved the levels of Zn, and in turn, SOD activity, which could be attributed to the antioxidant property of NA and Zn.
Results indicated a significant decrease in the activities of GPx and catalase in GIIa rats. These lower levels in GIIIa animals may be due to the Zn- and NA-deficient diet, as both Zn deficiency(Reference Oteiza, Clegg and Zago30) and NA deficiency(Reference Tang, Sham and Hui31) lead to the generation of OS. These activities were improved with increasing NA supplementation in the diet. NA supplementation alone(Reference Cho, Kim and Rodriguez-Iturbe32) or in combination with other factors(Reference Perumala, Shanthib and Sachdanandama33, Reference Salama, Nassar and Nafady34) has been shown to mitigate the up-regulation of oxidative and inflammatory systems with a decrease in NO, lipid peroxides and an increase in GSH and SOD levels.
In the present study, a significant increase in the lipid peroxidation level was observed in OS-treated and NA-deficient rats (GIIa), and supplementation of NA (GIIb and GIIc) resulted in a decrease in malondialdehyde levels. This may be due to increased hepatic Zn contents. The result seems to support the previous findings(Reference Parsons and Disilvestro25, Reference Oteiza, Olin and Fraga35, Reference Kraus, Roth and Kirchgessner36), wherein it has been suggested that rats fed on a Zn-deficient diet are more susceptible to peroxidative tissue damage. Also, the observed increase in lipid peroxidation was concurrent with a remarkable decrease in GSH levels in OS-treated rats as well as in non-OS-treated groups. Younes & Siegers(Reference Younes and Siegers37) have also observed that depletion of GSH enhances lipid peroxidation. GSH is a central constituent of antioxidant defence and acts as an essential cofactor for antioxidant enzymes such as GPx(Reference Kidd38). Under OS, GSH is consumed by GSH-related enzymes to detoxify peroxides in the water phase(Reference Cathcart39). Recently, Cortese et al. (Reference Cortese, Suschek and Wetzel40) demonstrated that Zn protects endothelial cells from H2O2-induced OS by increasing GSH biosynthesis. In the present study, the observed marked elevation in lipid peroxidation and depletion in GSH activity in GIIa animals suggests that depleted GSH stores may partly result in increased peroxidation, which are otherwise capable of moderating the amount of lipid peroxidation. However, NA supplementation to OS-treated Zn-deficient GIIb and GIIc rats significantly increased the levels of GSH compared with GIIa animals. NADPH is an essential coenzyme of the GSH reductase, which is involved in the reduction of oxidised to reduced GSH. Treatment with excess NA in GIIc showed near-normal levels of GSH, which may be conceived as the effect of enhanced tissue Zn levels by NA, reduction in hepatic peroxidative damage followed by a respite in GPx activity, thereby leading to restoration of the GSH content. In GIII animals, GSH levels improved accordingly with increased dietary NA contents.
It is well known that the liver performs an important function in the short-term regulation of trace-element metabolism(Reference Failla and Kiser41). Through its myriad biological functions, Zn plays an important role in the therapy of several liver diseases and has been shown to attenuate or protect against a variety of hepatotoxins such as carbon tetrachloride, bromobenzene and several metals(Reference Liu, Kershaw and Klaassen42–Reference Afonne, Orisakwe and Ndubuka44). The OS pre-exposure study indicated a significant depression of hepatic Zn content after Zn deficiency and OS treatment. However, NA treatment from GIIa to GIIc rats raised the hepatic Zn levels compared with the reference levels of GI rats. Interestingly, administration of NA to Zn-deficient GIII rats without OS treatment had not improved the hepatic Zn content to that extent, indicating the specific role of OS for its interaction with NA and Zn. Thus, in OS pre-exposure, it was found that NA enhanced the hepatic Zn level, which in turn might have resulted in hepatoprotection against OS-induced damage.
The interaction between Zn and NA is bidirectional as supplementation of any one improves the metabolism of the other. Vannucchi et al. (Reference Vannucchi, Kutnink and Sauberlich45) had reported that in NA-deficient rats, Zn repletion caused activation of NA metabolism. Similar results were obtained in another study from our laboratory(Reference Tupe, Tupe and Tarwadi15).
Exogenously added NA has been shown to markedly increase NAD levels in mammalian tissues including liver, kidney and heart(Reference Jackson, Rawling and Roebuck17) and concomitantly resulting in the reversal of OS-induced cell injury in kidney epithelial HEK 293 cells(Reference Hara, Yamada and Shibata3). Maiese et al. (Reference Maiese, Chong and Hou2) had reviewed the role of nicotinamide as an antioxidant and different unique cellular pathways involving nicotinamide that determine cellular longevity, cell survival and unwanted cancer progression. In vitro Zn uptake by human erythrocytes has been studied using blood samples of ten healthy subjects. It has been found that 8 mm-NA or NADPH increased 65Zn uptake by 38·9 and 43·1 % in the fasting, and by 70·9 and 28·1 % in postprandial conditions(Reference Agte, Paknikar and Chiplonkar14). When mice were fed with NA-deficient, -adequate and -excess synthetic diets for 4 weeks, it has been observed that, in comparison with the NA-deficient diet, percentage of Zn absorption, intestinal Zn, percentage of Hb and hepatic Fe increased significantly under NA-adequate and -excess conditions(Reference Agte, Paknikar and Chiplonkar14). The present study imparts another dimension to the antioxidant role of NA through the improvement in hepatic Zn uptake.
The results of all parameters signify that NA supplementation is more beneficial under the pre-exposure condition, as it assures more favourable circumstances to combat oxidative injury by enhancing Zn uptake under Zn deficiency. Hence, NA supplementation can be a better treatment strategy rather than prophylaxis.
In conclusion, dietary NA supplementation improves Zn uptake, acts as an antioxidant along with Zn and results in hepatoprotection against OS-induced damage in rats. The results collectively suggest that NA supplementation may have a therapeutic potential in the treatment of Zn deficiency-related complications and oxidative liver damage.
Acknowledgements
R. S. T. thanks the University Grants Commission, India, for a Senior Research Fellowship. All authors declare that they have no commercial/financial conflict of interest. R. S. T. and S. G. T. were involved in the study design, data collection, data analysis, data interpretation, literature search and manuscript preparation. V. V. A. was involved in the study design, data analysis, data interpretation, literature search, manuscript preparation and review of the manuscript.







