



Introduction
Progressive supranuclear palsy (PSP) is a neurodegenerative disorder that causes a mixed constellation of motor and behavioral symptoms associated with the deposition of pathological 4-Repeat tau protein following a sequential distribution in the brain but showing distinct regional predominance of pathology based on the clinical phenotype. Reference Kovacs, Lukic and Irwin1 To date, there are no proven therapies to slow or prevent disease progression, and management is symptomatic and supportive but limited in efficacy.
PSP is uncommon and affects 5 in 100,000, Reference Giagkou, Höglinger and Stamelou2 although postmortem studies suggest that it may be more common than currently appreciated. Reference Golbe3,Reference Respondek, Kurz and Arzberger4 Making a diagnosis of PSP is based on clinical criteria. As such, expertise in recognizing and managing the disease is generally within the scope of tertiary centers in Canada with specialized movement disorder or behavioral neurology clinics. Even in such referral centers, care can be fragmented due to the requirement of multiple sub-specialties to address the multiple systems affected by PSP. Clinical research is also challenging due to diagnostic difficulties and rapid clinical deterioration. An important contribution to this problem is the relatively recent recognition of clinical phenotypic heterogeneity and the relatively poor diagnostic accuracy in life, apart from the more classical cases of the Richardson syndrome (PSP-RS) variant. Reference Höglinger, Respondek and Stamelou5,Reference Ali, Botha, Whitwell and Josephs6 The commonest of these non-RS phenotypes include variants dominated by corticobasal syndrome (PSP-CBS), parkinsonism (PSP-P), frontal behavioral abnormalities (PSP-F), progressive freezing of gait (PSP-PFG), speech and language dysfunction (non-fluent aphasia; PSP-SL), and prominent early postural instability (PSP-PI). The related 4R-tauopathy corticobasal degeneration is equally heterogeneous in presentation and even more often misdiagnosed. Reference Saranza, Whitwell, Kovacs and Lang7,Reference Armstrong, Litvan and Lang8 To our knowledge, there exists no program in Canada that focuses on PSP, embedding clinical and basic research in clinical care. Although there are specific programs for atypical parkinsonism or frontotemporal dementia in tertiary referral centers in the US, there are very few that have been specially endorsed by the CurePSP Foundation as Centers of Care (which includes a high commitment to multimodal and standardized clinical assessments and provides interdisciplinary care) and none in Canada that are dedicated to the combination of clinical and basic research.
To improve management and clinical research in PSP and related tauopathies, with the support of the Rossy Foundation, we have recently established a dedicated clinical research program with research embedded in care, the Rossy PSP Centre. We present here the design and operationalization of the program for the diagnosis, long-term follow-up, and development of a research cohort with careful phenotyping of patients with probable PSP and CBS for improved clinical-pathological correlations. Additional goals are the possible description of new phenotypes and the discovery of new fluid and tissue biomarkers with the support of postmortem assessment of patients enrolled in the research program.
Methods
Rossy PSP Centre’s Program Design
Aim and Mission
A Mission statement for the Rossy PSP Centre at the Toronto Western Hospital (TWH), University Health Network (UHN), was formulated: To establish a world-class clinical and basic research program in PSP and related Atypical Parkinsonian disorders with the primary goal of advancing the understanding of these complex neurodegenerative diseases and eventually contributing to the development of effective disease-modifying therapies. Critical components of the program are the provision of best available care and embedding research in clinical care, availing patients of the latest research studies including clinical trials of experimental therapies, the recruitment of bright young investigators to the field as fellows in training and an open and sharing approach to collaborative research with the internationally renowned members of the Program’s Scientific Advisory Board (SAB) and other experts throughout the world.
In terms of program design, a Scientific Advisory Committee and an external advisor have proven crucial to shape the consistency of clinical and research goals and their integration with similar past and ongoing international initiatives. Members of the SAB and external advisors include the following experts: A. Boxer (US), L. Golbe (US), G. Hoglinger (Germany), I. Litvan (US), I. Mackenzie (Canada), H. Morris (UK), and M. Samuels (US). Recently, the program has been granted Center of Care status by the CurePSP Foundation.
Goals and Design
The program’s clinical goal is to provide diagnostic and management recommendations to the patient, the family, and to the referring physician. In case the patient is under the care of a general neurologist or other movement disorders/behavioral neurology specialist, the program encourages the patient to retain this care while providing recommendations on treatment and referrals to additional professionals required from the allied-health team (i.e., speech/language pathologists, occupational therapy, physiotherapy, urology, and other pertinent medical specialties). The program’s scientific goal is to conduct a prospective, longitudinal observational study of the clinical presentation and progression of PSP and other possible tauopathies with deep phenotyping of patients using a variety of clinical assessments, imaging studies (magnetic resonance imaging (MRI), positron emission tomography) and biological samples (e.g., blood, saliva, skin biopsy and skin oil, cerebrospinal fluid (CSF)). The visit schedule with the list of scales and questionnaires administered are presented in Table 1. Funding for the Rossy Centre was received in 2019, and the clinical arm of the program was initiated in October of that year (following hiring of a fellow, research assistant, and research coordinator). In consultation with other programs in Europe and the United States, the faculty established the formal clinical evaluation tools/protocols during the planning phases and applied these immediately on initial patient intakes using paper documentation while plans were being made for the electronic database. The final choice of the database vendor was made over the first year of the clinical program, and once this was finalized, all the clinical data were subsequently entered. In parallel with the clinical assessments and care provided in the program, research ethics board submissions were made for the ongoing use of the clinical data for research purposes as well as collection of biological samples for a variety of research purposes including biomarkers and genetics. The time required from the initial planning stages to the current fully functioning clinical program has been approximately 2.5 years.
Table 1: Visit schedule with the list of scales and questionnaires administered

ARTFL, Advancing Research and Treatment in Frontotemporal Lobar Degeneration; LEFFTDS, Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects; MDS-UPDRS, Movement Disorders Society-Unified Parkinson’s disease Rating Scale; MoCA, Montreal Cognitive Assessment; CGI, clinical global impression; TorCA, Toronto Cognitive Assessment; PDQ, Parkinson’s disease questionnaire; EQ-5D-3L, EuroQoL, 5-dimension 3 levels; CBS, corticobasal syndrome; RSMS, The Revised Self-Monitoring Scale.
Clinical Assessments
Initial visits have a duration of 120–180 minutes including a first clinical assessment by a neurology fellow, a discussion with the faculty physician, and a final encounter/interview with the research coordinator where the scheduling of further appointments and completion of additional evaluations are done. Follow-up visits (between 90 and 120 minutes) are held every 6 months during the first year and then every 12 months. Their main goal is the collection of clinical data regarding the progression of the disease. As part of follow-up, a virtual interview is held by the research assistant (see below Pre-clinical interview and questionnaires). Clinical data collections have been chosen to harmonize with those obtained by other large consortia on PSP to facilitate data sharing.
The clinical diagnosis is made by application of the MDS-PSP criteria, Reference Höglinger, Respondek and Stamelou5 and clinical features are documented in specific forms as well as the primary, secondary, and tertiary most likely diagnoses together with the clinician’s confidence level in each of these (percentage). The flow chart presented in Figure 1 shows the path of each patient from referral to the program through follow-up options and inclusion in different clinical research opportunities offered.

Figure 1: Patient’s path through clinical care and research within the Rossy Program.
Pre-clinical Interview and Questionnaires
The MDS-UPDRS part II, Clinical Dementia Rating scale, and the Epworth sleepiness scale are assessed virtually in a pre-clinical meeting with patient and relative. In addition to these, scales of quality of life, behavioral symptoms, and information about family history, previous medication use, and occupational exposures are collected during this visit (see Table 1).
Biobanking for Detection and Development of Biomarkers
Biofluid Specimen and Skin Biopsy
All patients are asked to participate in biobanking, which includes blood, saliva, skin (3–5 mm punch biopsies of cervical skin), and cerebrospinal fluid.
Genomic Analyses
PSP is genetically heterogeneous; however, this has been investigated in only a few studies mainly limited to cohorts of European ancestry. The strongest PSP-association signal is reported for variants in MAPT mapped to the inversion region linked to the H1/H2 tau-haplotype. Reference Hoglinger, Melhem and Dickson9 Therefore, all study participants are assessed for H1/H2 haplotype. Notably, genome-wide association studies of Parkinson’s disease (PD) have also detected a highly significant association with the H1-haplotype of MAPT, Reference Spencer, Plagnol and Strange10,Reference Nalls, Pankratz and Lill11 pointing to an overlap in disease mechanisms between PD and PSP (also clinically evident in PSP-P patients). Thus, study participants are also assessed for mutations in PD-related genes. Genetically characterized autopsy tissues could reveal new gene candidates derived from the neuropathologically well-characterized cases. Hence, our priority is to start with a genetic study of pathologically confirmed cases of PSP and other tauopathies and contrast these with other neurodegenerative disorders such as PD, multiple system atrophy, dementia with Lewy bodies, and Alzheimer’s disease. During the first 2 years of the Rossy program, we generated a DNA collection from 384 well-characterized autopsy cases (collected before the establishment of the Rossy Centre). For the ongoing genetic analysis of this brain cohort, we selected the most modern genome-wide Illumina array tailored to study neurological diseases (GDA-NeuroBooster Bead Chip 8+v1.0). Specifically, the Neuro Booster array is based on the global diversity array backbone that provides genotypes for about two million variants (https://www.illumina.com/products/by-type/microarray-kits/infinium-global-diversity.html) including up to 95,000 custom content variants relevant to neurological disease (https://github.com/GP2code/Neuro_Booster_Array). Keeping in mind that novel genetic variations cannot be detected by the Neuro Booster array, we have also established a collaboration with Dr. Z. Gan-Or from McGill University (Montreal, Canada) who is currently genotyping our brain DNA collection using next-generation targeted sequencing of 51 parkinsonism-related genes, including the MAPT locus. In addition to these prioritized genetic studies, we will also be conducting epigenetic studies given the influence of DNA methylation (DNAm) at CpG-sites as a key epigenetic regulator of gene expression that can influence clinical presentation, including age of onset and rate of progression (see Discussion).
Brain Donation
The Rossy program has been associated with the launching of a more active neurodegenerative diseases brain donation program offered to all patients and families in the movement disorders and behavioral neurology programs at TWH. The option of brain donation is also discussed in a supportive, respectful fashion with all patients expressing an interest in considering medical assistance in dying (MAID) and this has permitted a very short death to autopsy time (typically 6 hours or less) that has made it possible to successfully culture neurons and glia for future research studies. We have established standardized protocols for handling, processing, staining, and storing of brains and necessary infrastructure supporting the neuropathology laboratory for the prospective brain collection. The strategy includes preparing formalin-fixed paraffin-embedded tissue samples from both hemispheres, including corresponding regions that are sampled for deep freezing (−80 Celsius degrees) for biochemical, seeding assay, Reference Martinez-Valbuena, Visanji and Kim12 transcriptomic and genomic/epigenomic studies. In addition, the laboratory is prepared to process double or half hemispheric large blocks. In addition to detailed written neuropathology reports, computer-based data collection comprises a detailed diagnostic sheet and an extensive evaluation sheet to document various pathologies including immunoreactivities using immunostaining for neurodegenerative disease-related proteins, the latter used to generate heat maps of pathologies. Reference Couto, Martinez-Valbuena and Lee13,Reference Di Luca, Slow, Martinez-Valbuena, Lang and Kovacs14 Establishment of postmortem MRI imaging has also been organized. Our approach to brain sampling and evaluation will be reported in a separate paper. Finally, we have established a preliminary agreement with CurePSP to fund the harvesting, transportation, and evaluation of PSP brains from other centers in Canada.
Staff Working at the Rossy Program
The human resources in the program include one program coordinator, one research assistant, and at least five physicians with neurology specialty: three movement disorders sub-specialists, one behavioral-cognitive sub-specialist, one neuropathology specialist (also movement disorders neurology specialist) and a designated Rossy movement disorders fellow as well as other movement disorders/behavioral neurology fellows seeing patients in the program on a rotational basis.
Ethical Review and Approval
The entire clinical and research protocol was approved by the Research Ethics Board of the UHN, and patients and caregivers sign an approved informed consent form to participate in research within the Rossy program.
Data Collection and Storage
The information collected at each visit is uploaded into the DAta Driven Outcome System (DADOS) digitized platform (Techna Institute, UHN, Toronto, Canada). DADOS is a platform used by several research clinics affiliated to the UHN. This provides a set of digitized forms organized in a structure of folders per visit and per type of administration (self-answered by patient/caregiver, administered by research coordinator or by physician). They can be accessed online by the staff of the program who are given a password and username. Pictures of source data can be scanned and uploaded. Collated data can be analyzed at any time. Data storage in DADOS uses different formats including text, numeric, image counts with a password-protected access. The actual programmed PSP DADOS database will be shared with other academic clinics wishing to establish a comparable clinical research program. Use of the DADOS platform will facilitate sharing of relevant clinical assessments, environmental history, and cognitive-behavioral symptoms data to promote data collaboration with similar academic clinics, clinical registries, and research initiatives on PSP. Responsibility of protecting patient data throughout any sharing process will be assumed by the PI/applicant of the proposal/request.
Results
Referrals have been accepted, both from external and internal physicians to UHN. The combined pool of internal referrals came from the Movement Disorders Clinic that has followed about ∼300 patients with PSP since 2011 and the Memory Neurology Clinic of the TWH with another ∼200 patients in follow-up. However, considering the median survival of 7.8 years and a mean disease duration at the time of referral of about 5.6 years, many of these patients were either too advanced to attend the clinic or lost to follow-up by the time the Rossy program launched, resulting in internal referrals involving patients seen for the first time after 2016. In 27 months, between October 2019 and December 2021, a total of 132 patients were screened. Criteria for clinical PSP were fulfilled by 110 patients. Details on the number of follow-up visits and excluded patients are provided in Figure 2. Demographics, clinical features, and predominant phenotype of the probable PSP are shown in Table 2. Initial dominant complaint at presentation was postural instability and falls in 45%, cognitive-behavioral changes in 22%, Parkinsonism in 9%, and tremor or myoclonus in 7%, as well as oculomotor abnormalities in 7% and bulbar symptoms in 7%.
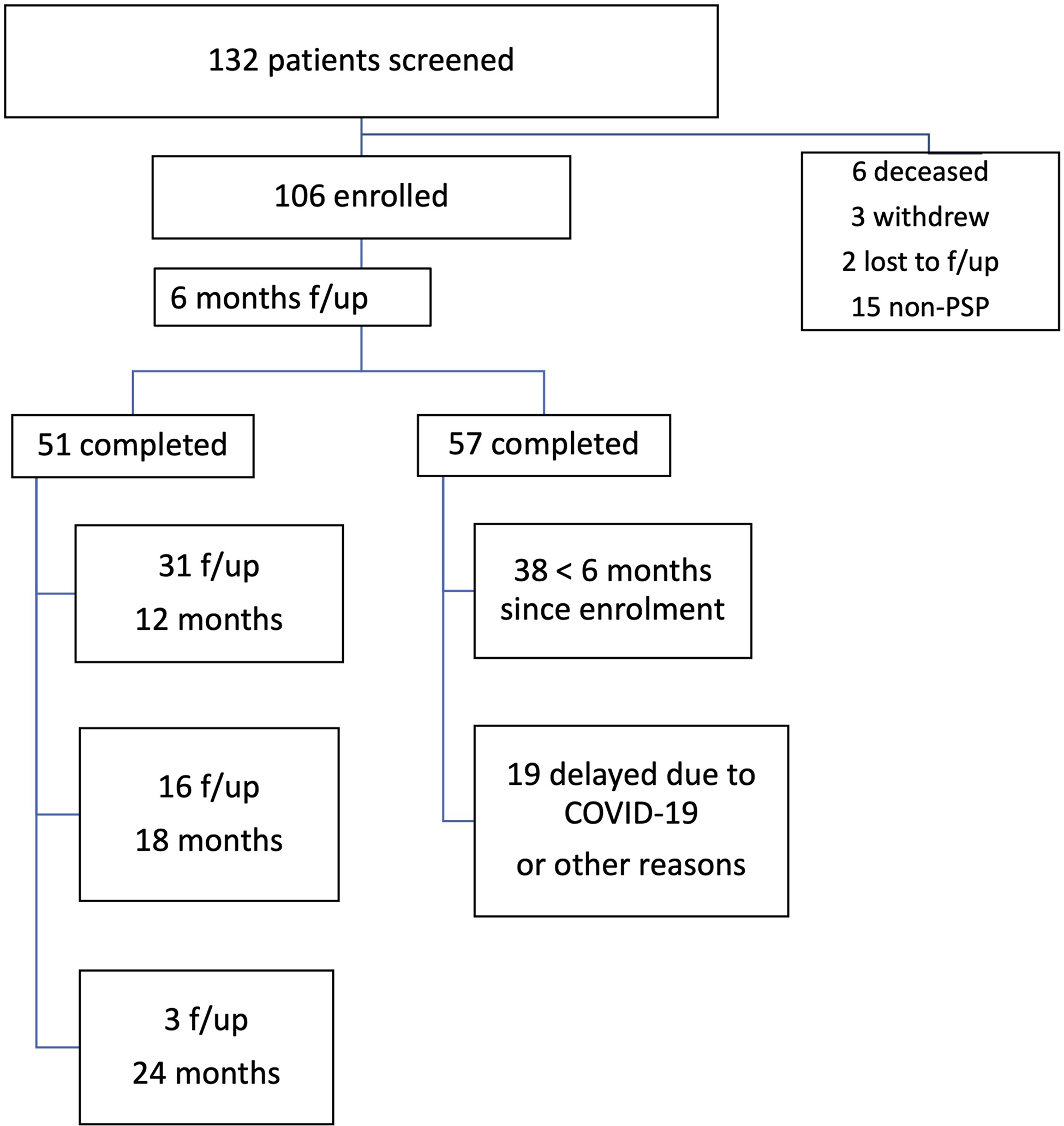
Figure 2: Enrollment and follow-up visits.
Table 2: Results of the screened patients’ demographics, disease features, and phenotypes
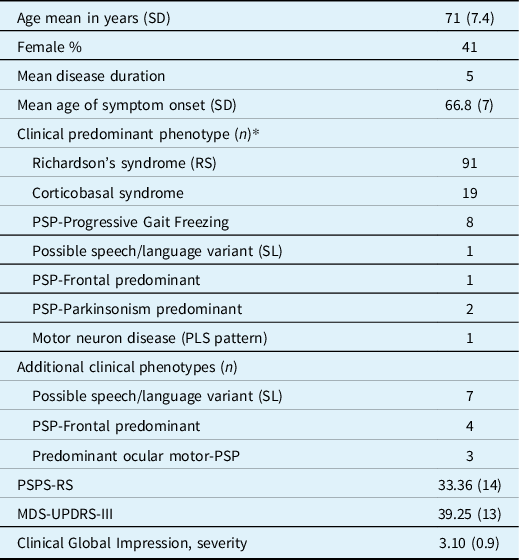
SD, standard deviation; Age, disease duration and age of symptoms onset in years; PLS, primary lateral sclerosis; PSP-RS, progressive supranuclear palsy rating scale; MDS-UPDRS-III, Movement disorders society endorsed Unified Parkinson Disease rating scale, part 3.
* Twenty-seven patients fulfilled criteria for more than one variant (listed as primary and secondary phenotypes), and all of them later developed Richardson syndrome: 10 patients with CBS manifested additional RS or SL/progressive aphasia variants, 15 patients had a non-Richardson and Richardson phenotype, and two patients fulfilled criteria for three variants.
In terms of management, diverse symptomatic treatments were attempted for motor and non-motor symptoms. Levodopa use was documented in 74 patients (mean dose 500–600 mg per day) and amantadine in 54 patients (mean dose 250 mg per day). Response was defined based on patient’s report as (i) mild subjective benefit in either balance, stiffness, or general wellbeing including more alertness; (ii) moderate benefit of these features but no impact in daily living functionality; (iii) considerable improvement in balance, stiffness, gait, or cognition and with a relevant impact in daily living functionality; (iv) no improvement or worsening of balance or overall function. Moderate and persistent subjective response to levodopa was recorded in less than 25% of the patients without a clear change in motor signs on examination. In those who reported response to amantadine, it was generally mild (less than 40% benefit in motor and cognitive symptoms) and with a duration shorter than 2 months. In addition, botulinum toxin injections were indicated for the treatment of dystonia in patients with disabling blepharospasm/eyelid-opening apraxia (10 patients, response rate 70%), limb dystonia with pain or hygiene limitations in upper extremities (3 patients, response rate 100%), or foot dystonia interfering with walking (3 patients, response rate 33%). Sialorrhea was treated with atropine 1% drops, ipratropium bromide 0.03% spray, or botulinum toxin injections to the parotid and submandibular salivary glands (seven patients, response rate 85%).
Treatment of non-motor symptoms included the management of mood/behavioral and sleep issues. For these, pharmacological measures included selective-serotonin reuptake inhibitors drugs for symptoms of depression, anxiety, and emotional lability and irritability in about 40% of patients, and agents such as melatonin, and mirtazapine for insomnia in 14%. One patient with disabling pseudobulbar affect obtained marked benefit from the combination of dextromethorphan and quinidine prepared by a local pharmacy. Non-pharmacological treatment included periodic referral to allied-health specialists: speech/language pathology every 12 months in the 91 patients with dysphagia, and for communication issues related to dysarthria or language impairment; occupational therapy in 91 patients to address home safety and walking aid recommendations; physiotherapy for deconditioning, falls prevention and for range of motion in the case of immobility. Palliative care referral for patients in later stages of the disease is done by the social worker of the Memory clinic (five done until this time) or through the patient’s family physician.
Since the clinic was opened in Oct 2019, 7 of the 110 assessed patients have died; all of them have donated their brains for research including three following medical assistance in dying (MAID). This represents 10% of the total of 70 brain donations received to the whole Brain donation program during that time.
The Rossy Centre has a policy completely supportive of data sharing whenever requested by centers with shared and aligned research goals and reasonable justification is provided. All data collected and analyzed will be treated as confidential and the potential identifying information removed and anonymized. Standard operating procedures will be instated for each external data request and signature of a data sharing agreement between institutions.
Discussion
This article summarizes the design and first 2 years of experience of the Rossy PSP Centre at the Movement Disorders Clinic of the TWH. The goals of the program include improving the diagnosis, treatment, and quality of life of people with PSP and related disorders and to advance both clinical and basic research in this group of inexorably progressive and inevitably fatal neurodegenerative diseases. A total of 132 patients have been screened in 27 months, and about a hundred have been enrolled with various clinical presentations fulfilling MDS criteria of possible or probable PSP, and over 50% have already completed a first follow-up visit despite the intervening COVID pandemic. It is important to note that during this time, several research initiatives derived from this cohort have been conducted including epidemiological study of the cohort, the development of reliable diagnostic biomarkers, Reference Martinez-Valbuena, Visanji and Olszewska15 testing and validation of current diagnostic criteria, evaluation of a virtual version of the PSP Rating Scale, Reference Wills, Pantelyat and Espay16 and the description of a “protracted course PSP.” Reference Couto, Martinez-Valbuena and Lee13 An evaluation of the response to pharmacologic and non-pharmacologic treatments is under preparation for peer-reviewed publication.
The strengths of the program are its design utilizing the most recent clinical criteria and the deep phenotyping of the patients. An important goal is that increasingly larger numbers of patients will be seen at earlier disease stages with a broader representation of PSP variants. This is an important goal since a strong bias towards over-representation of the most common Richardson’s syndrome (RS) variant exists in previous clinical and clinical-pathological studies. Reference Golbe3 For example, RS accounted for 53% of an initial series that clustered symptoms in two variants (RS and PSP-Parkinsonism) Reference Williams, Holton and Strand17 but a series containing patients with the six currently described variants showed an incidence of only 24% of RS at presentation. Reference Respondek, Kurz and Arzberger4 Although our preliminary results show that 91 patients fulfilled criteria of a Richardson syndrome, this number includes those patients who developed this even after having a different presentation at onset. Following the recommendations of use of the MDS-PSP criteria, Reference Grimm, Respondek and Stamelou18 RS is the most prevalent and with highest positive predictive value (“probable” PSP) compared to other phenotypes such as speech/language variant at onset. To mitigate for this finding, the Rossy program includes careful historical documentation of primary, secondary, and tertiary clinical phenotypes for each patient. Table 1 shows the frequency of the secondary phenotypes; many cases did not start with a RS but, as is typically the case, later evolved to this but before the initial visit to the Rossy program. This decrease in phenotypic diversity has been reported with other clinical and pathologically confirmed cohorts. Reference Respondek, Kurz and Arzberger4,Reference Sánchez-Ruiz de Gordoa, Zelaya and Tellechea-Aramburo19 Further attempts to increase a broader representation of PSP variants include screening of all referrals to our movement disorders and behavioral neurology programs for possible PSP variants (e.g., behavioral variant frontotemporal dementia, non-fluent primary progressive aphasia, CBS, primary freezing of gait), and these are directed to the Rossy program for initial assessment. Finally, we expect that as the general awareness of this new program in the neurologic community increases this will enhance referral of a broader clinical profile.
The longitudinally collected data will provide insight into the diagnosis and management of PSP and allow the creation of trial-ready cohorts of patients to be fast-tracked to participate in future trials of experimental disease-modifying therapies. For example, the variability in clinical presentation of PSP could be attributed to genetic polymorphisms in SLC2A13 Reference Jabbari, Koga and Valentino20 and TRIM11 loci, Reference Jabbari, Woodside and Tan21 in agreement with our recent study suggesting that a longer PSP duration might be related to homozygosity for the rs564309-C allele at TRIM11. Reference Couto, Martinez-Valbuena and Lee13 The catalog of significant findings by genome-wide association studies Reference Buniello, MacArthur and Cerezo22 includes 35 common variants, the most consistent being the association of PSP with the MAPT gene variant rs807072 Reference Hoglinger, Melhem and Dickson9 and the H1/H2 tau-haplotype. Genetic investigations of autopsy cases have been prioritized during the first 2 years of the program, and in a second stage, epigenetic factors such as DNA methylation regulating gene expression and potential prognostic biomarker Reference Xi, Zhang and Bruni23,Reference Zhang, Ferrari and Tartaglia24 will be studied in variants of PSP as we have tested in other neurodegenerative diseases. Reference Zhang, McKeever and Xi25,Reference Zhang, Tartaglia and Moreno26 The results of these studies may assist in explaining clinical heterogeneity in PSP and may provide important considerations in the design of future disease-modifying clinical trials.
The new staging of pathology of PSP and the continuous search for diagnostic biomarkers (in blood and plasma, skin, and CSF), including seeding amplification assays, Reference Martinez-Valbuena, Visanji and Olszewska15,Reference Tarutani, Miyata and Nonaka27 exosomes derived from neurons and glia, Reference Ngolab, Trinh and Rockenstein28–Reference Stuendl, Kraus and Chatterjee31 as well as studies of tau using cryo-EM, Reference Shi, Zhang and Yang32 guarantee advances in our knowledge over the next few years and will contribute to the development of biomarkers for therapy trials. The brain donation program provides feedback for the clinical phenotyping, including understanding protracted course Reference Couto, Martinez-Valbuena and Lee13 and rapidly progressive forms of PSP, allows comparative studies with neuroimaging, and provides an invaluable resource for tissue-based research in PSP.
Acknowledgements
The authors would like to thank the Rossy Foundation and family for their generous support and the patients and their families involved in the program. The funding has supported a dedicated fellow, full-time research assistant, part-time research coordinator, programming, and ongoing support of the database as well as ongoing clinical and basic research projects. Initial funding has been provided for a 10-year period.
Statement of Authorship
B.C.: Data curation, Writing-Original draft preparation.
S.F.: Conceptualization; Supervision; Data curation, Writing – Reviewing and Editing.
M.C.T.: Conceptualization; Supervision; Data curation, Writing – Reviewing and Editing.
E.R.: Conceptualization; Writing – Reviewing and Editing.
J.A.: Data curation, Writing – Reviewing and Editing.
P.B.: Data curation, Writing – Reviewing and Editing.
G.G.K.: Conceptualization; Supervision; Data curation, Writing – Reviewing and Editing.
A.E.L.: Conceptualization; Supervision; Data curation, Writing – Reviewing and Editing; Funding acquisition.
Conflicts of Interest
The authors deny any conflict of interest regarding the data described in this article.
Disclosures
Dr. Lang has served as an advisor for AbbVie, AFFiRis, Alector, Amylyx, Biogen, BioAdvance, BlueRock, BMS, Denali, Janssen, Jazz, Lilly, Novartis, Paladin, Retrophin, Roche, Sun Pharma, and UCB; received honoraria from Sun Pharma, AbbVie and Sunovion; received grants from Brain Canada, Canadian Institutes of Health Research, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, Parkinson Foundation, Parkinson Canada, and W. Garfield Weston Foundation; received publishing royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press. Dr. Tartaglia has served as an advisor for Denali and received grants from the Canadian Institutes of Health Research, and National Institute of Neurological Disorders and Stroke. Dr. Fox receives clinic support from the Edmond J Safra Foundation for Parkinson’s Research; Parkinson Foundation; research funding from Michael J Fox Foundation for Parkinson Research, NIH (Dystonia Coalition); Parkinson Canada; honoraria from the International Parkinson and Movement Disorder Society; consultancy/speaker fees from Alexion, Bial, Pharma 2B, Sunovion, and Paladin. Royalties from Oxford University Press. Dr. Kovacs receives funding from the Rossy Family Foundation and Edmond Safra Philanthropic fund; received funding from Michael J. Fox Foundation, Parkinson Canada and Canada Foundation for Innovation; publishing royalties Wiley, Cambridge University Press, and Elsevier; previously received consulting fees from Biogen; declares a shared patent for 5G4 synuclein antibody with Roboscreen GmbH. The authors BC, PB, JA, and ER have nothing to declare.





 Open access
Open access