



Introduction
Tuberous sclerosis complex (TSC) is a rare, autosomal-dominant disorder characterized by hamartomas in multiple organs. Reference Islam1 It affects an estimated 1 in 6,000 to 10,000 individuals at birth Reference OʼCallaghan, Shiell, Osborne and Martyn2 and is caused by germline pathogenic variants in the tumor suppressor genes tuberous sclerosis complex 1 (TSC1) and tuberous sclerosis complex 2 (TSC2), which function as part of the mammalian target of rapamycin (mTOR) pathway to regulate cell growth, size and proliferation. Reference Randle3 De novo pathogenic variants are identified in two-thirds of individuals with TSC, while the remainder of variants are inherited. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4
Individuals with TSC often show multiple organ system involvement, including the brain, skin, kidneys, heart, lungs and eyes. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4 Nearly all patients with TSC present with skin manifestations, including hypomelanotic macules, facial angiofibroma and shagreen patches. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4 Central nervous system involvement is seen in 90% of patients with TSC, including subependymal nodules (SEN), cortical tubers and subependymal giant cell astrocytoma (SEGA). Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4 Furthermore, 80% of individuals with TSC will have a renal lesion by 10.5 years of age, Reference Ewalt, Sheffield, Sparagana, Delgado and Roach5 including benign angiomyolipoma and renal cysts. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4 Neurological and renal malignancies are the leading causes of death in TSC patients. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4
A clinical diagnosis of TSC is made in patients showing two major characteristics or one major characteristic and two minor characteristics. Reference Northrup, Aronow and Bebin6 In addition, a pathogenic variant in TSC1 or TSC2 is sufficient for a diagnosis of TSC. Reference Northrup, Koenig, Pearson, Au, Adam, Everman, Mirzaa, Pagon, Wallace, Bean, Gripp and Amemiya4 Although both genetic and clinical criteria are used to diagnose TSC, wide phenotypic variability often complicates the diagnosis, and 10%–25% of individuals with TSC do not have a germline pathogenic variant identified by regular genetic testing, referred to as no mutation identified (NMI). Reference Northrup and Krueger7 Due to wide phenotypic variability, patient care and management vary depending on an individual’s clinical features.
Previous studies have reported that patients with TSC2 pathogenic variants usually present with a more severe phenotype (i.e., when compared to TSC1 and those with NMI), which consists of a greater number of tubers, earlier age of seizure onset and greater risk for intellectual disability. Reference Curatolo, Moavero, Roberto and Graziola8 Eighty percent of those affected by TSC are diagnosed in infancy or early childhood; therefore, some individuals have unrecognized manifestations and do not receive a diagnosis until adulthood. Reference Nathan, Burke, Trickett, Moss and Darling9 Recent studies involving patients with TSC have a strong focus on children, leaving a gap in knowledge in the clinical manifestations in adults with TSC. Furthermore, multisystem involvement in adults with TSC has yet to be reported in depth in the current literature, which brings the need to highlight how the progression or delayed diagnosis of TSC may affect the adult population. As a result, it is imperative to explore genotype-phenotype correlations to ensure structured management and surveillance are maintained with the variable phenotypic expression seen over time in adult patients with TSC. Reference De Sautu De Borbón, Guerra Vales and Lumbreras Bermejo10 In this study, we aimed to characterize the phenotypic and molecular characteristics of a Canadian adult TSC population to improve multidisciplinary care and adult patient management.
Materials and Methods
Participant Inclusion
We performed a retrospective chart review of adult TSC patients who were followed at the Fred A. Litwin Center for Genetic Medicine (University Health Network, Toronto, Canada) between January 1, 2001–August 10, 2020. Male and female patients ≥ 18 years of age with a clinical Reference Northrup, Aronow and Bebin6 or genetic diagnosis of TSC were reviewed; however, only individuals with a definitive diagnosis of TSC were included in the analysis. Data was collected through electronic patient records and paper charts, with information entered into a REDCap database. Approval was obtained from the Research Ethics Board at the University Health Network.
REDCap Database
The Research Electronic Data Capture (REDCap) database was developed as a secure, web-based platform for research teams to collect, store and disseminate project-specific information. Reference Harris, Taylor, Thielke, Payne, Gonzalez and Conde11 An internally housed REDCap database was constructed to collect patient medical history, family history and information relevant to the adult TSC experience. All patients were offered screening for possible TSC manifestations, per updated clinical surveillance guidelines. Reference Northrup, Aronow and Bebin6 Patients were typically offered a follow-up appointment every 12–16 months, where their interval medical history was reviewed and investigations were arranged.
The REDCap database included relevant information related to demographics, family history, genetic testing and TSC manifestations. TSC clinical features, such as neurological, skin, renal, eye, cardiac, respiratory, liver and reproductive systems were captured for analysis. In addition, behavioral information was also recorded, as was patient management information, such as imaging of affected systems and current/previous interventions. Finally, genetic testing results, including variant type, variant nomenclature, inheritance pattern and certainty of diagnosis, were recorded.
Genetic Testing
Of the 51 patients in our cohort, eight did not have genetic testing done (16%). The remaining 43 patients (84%) had genetic testing done by next-generation sequencing, sanger sequencing and multiligation-dependent probe amplification. Genetic testing results were classified according to the American College of Medical Genetics standards and include pathogenic, likely pathogenic and variants of unknown significance. Reference Hampel, Bennett, Buchanan, Pearlman and Wiesner12 All genetic test results were reviewed by the clinic’s Genetic Counsellor and Medical Geneticist. For our study, eligible patients were grouped according to their genetic testing results (see Fig. 1).

Figure 1: Categorization of 51 eligible patients into groups based on genetic testing results.
Statistical Analysis
The frequency of clinical manifestations was compared between the TSC1 and TSC2 groups, as well as between the TSC1 or TSC2 and the NMI group. R-studio (version 2022.7.1.554) was used to perform a chi-square test for the categorical data collected in this study. 13 P-values < 0.05 were considered statistically significant.
Results
Our cohort included 51 patients with a confirmed clinical diagnosis of TSC. The age range of the patients was 19 years to 66 years, with a median age of 32 years. Fifty-one percent (26/51) of the cohort were male and 49% (25/51) were female. Sixteen percent (8/51) of individuals had a TSC1 variant and approximately 35% (18/51) of patients had a TSC2 variant confirmed by genetic testing. In addition, four individuals had variants identified in both the TSC1 and TSC2 genes, and two patients had whole deletions of both the TSC2 and PKD1 genes. Twenty-two percent (11/51) of our cohort had NMI through germline genetic testing and an additional eight individuals did not have confirmatory genetic testing; however, they met the clinical criteria of TSC. Table 1 displays genetic testing results of patients included in the study, grouped by gene.
Table 1: Variants associated with TSC Canadian adults. TSC2 was the most frequent variant identified in this cohort. Reference sequences: NM_000368.5 (TSC1); NM_000548.5 (TSC2)
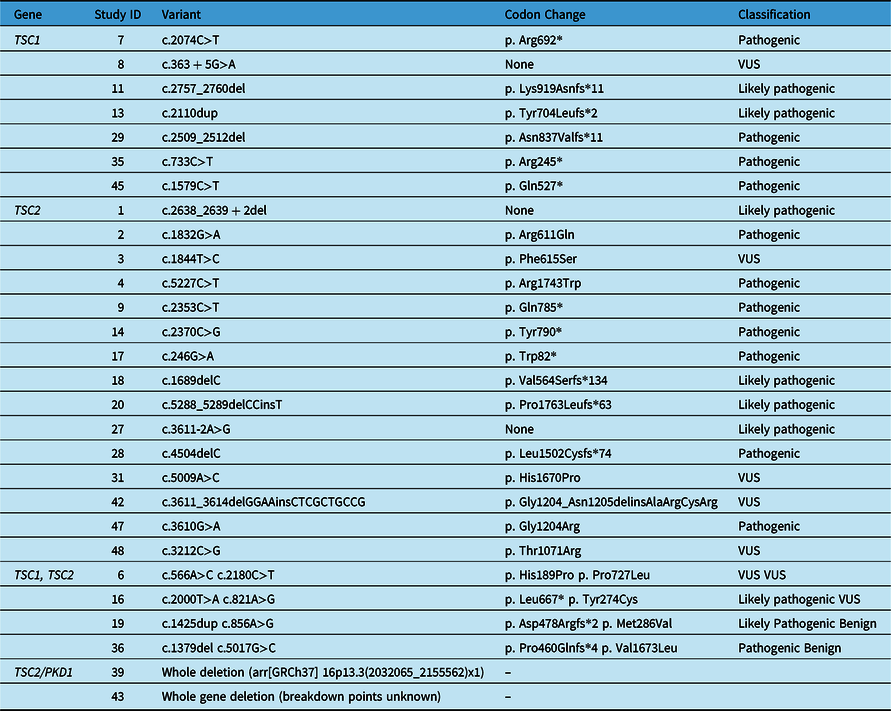
None = Splice site variant; VUS = Variant of unknown significance.
For the purpose of this analysis, we focused on the most prevalent systems affected: neurological, skin, renal, eyes, liver, cardiac and the lungs. Within our cohort, the most prevalent manifestations included: SEN, SEGA, cortical tubers, renal angiomyolipoma’s (AMLs) and skin features, such as hypomelanotic macules (Tables 2 and 3). Organ system involvement varied among patients within the cohort. The systems that were observed to be most affected in our cohort were the neurological, dermatological and renal systems. However, only the dermatological and cardiac manifestations had statistically significant differences when compared across the phenotypes (Table 2). Dermatological manifestations were present among all groups, with hypomelanotic macules being the most frequent feature. Other prevalent features include shagreen patches and facial angiofibroma. Cardiac rhabdomyoma was the most prevalent cardiac feature amongst our cohort and was recorded only in the TSC2 group, “other” variants and the NMI group.
Table 2: P-values yielded from chi-square test comparing frequency of manifestations between the TSC1 and TSC2 groups and comparing TSC1/TSC2 group with the NMI group. P-values < 0.05 are considered statistically significant

ASD=autism spectrum disorder; AML=angiomyolipoma; LAM=lymphangiomyomatosis; SEN=subependymal nodules; SEGA=subependymal giant cell astrocytoma; TAND=TSC-associated Neuropsychiatric disorder.
* indicates p-value<0.05; statistically significant,
Table 3: Clinical manifestations present in an adult TSC cohort of 43 patients. Manifestations in the brain, kidney and skin were the most frequently reported. NM_000368.5 (TSC1); NM_000548.5 (TSC2)
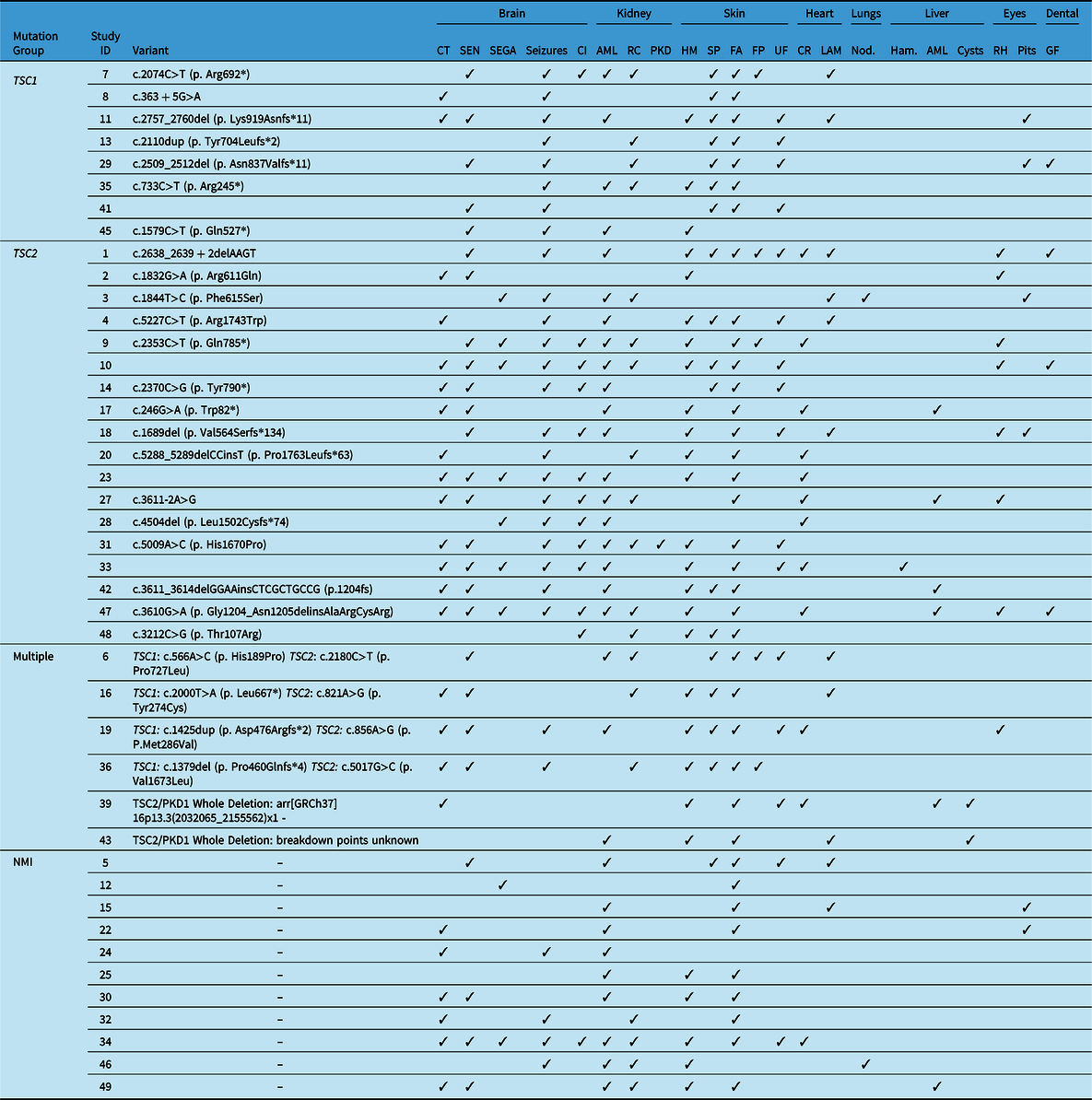
AML=angiomyolipoma; CI=cognitive impairment; CT=cortical tubers; CR=cardiac rhabdomyoma; FA=facial angiofibroma; FP=forehead plaque; GF=gingival fibroma; Ham=[liver] hamartoma; HM=hypomelanotic macules; LAM=lymphangioleiomyomatosis; NMI=no mutation identified; Nod=[liver] nodules; RC=renal cysts; RH=retinal hamartoma; PKD=polycystic kidney disease; SEN=subependymal nodules; SEGA=subependymal giant cell astrocytoma; SP=shagreen patches; UF=ungual/peri-ungual fibroma.
Although there were no statistically significant differences detected between the other systems affected in TSC and genotype, some notable findings were observed. TSC-associated neuropsychiatric features (TAND) varied among the different groups, with behavioral disturbances and cognitive impairment observed most frequently. Of the individuals with a contiguous mutation in TSC2/PKD1, interestingly both presented with AMLs and cysts, which led to hypertension. mTOR treatment was given in 35% (18/51) patients; 16 patients were given Everolimus and two patients were given Sirolimus to manage systemic features but with an emphasis on renal manifestation management.
A summary of the observed frequency of clinical manifestations and statistical significance of our analysis based on chi-square tests between TSC1 and TSC2 and between the TSC group with the NMI group can be found in Table 2. A summary of clinical manifestations and genetic testing results can be seen in Table 3.
Discussion
Our study aimed to examine genotype and phenotype relative frequencies in adults with TSC. Our cohort consisted of 51 adult patients, with an age range of patients 19 to 66 years of age, consisting of 51% (26/51) males and 49% (25/51) females. The most prevalent manifestations in our cohort included: SEN, SEGA, cortical tubers, renal AMLs and skin features, such as hypomelanotic macules. We observed that approximately 16% of individuals had a TSC1 variant and 35% presented with a TSC2 variant. This finding is consistent with previous reports from the literature, showing that TSC2 variants are more common than variants in TSC1. Reference Rosset, Netto and Ashton-Prolla14
TSC2 variants are associated with a more severe phenotype with age; Reference Kothare, Singh and Chalifoux15 in our cohort, those with TSC2 variants presented with SEN, SEGA and cortical tubers more frequently than those with TSC1 variants. However, due to the lack of statistical significance, our data does not confidently support previous studies showing that SEN and SEGA are more common in patients with a TSC2 variant. Reference Kothare, Singh and Chalifoux15 This is likely secondary to small sample size and lack of power to detect such differences. In reviewed literature, however, tubers often did not differ between the two groups. Reference Kothare, Singh and Chalifoux15 In addition, TSC2 variants are associated with higher risk of intellectual disability. Reference Alsowat, Whitney and Hewson16 Our data was consistent with these findings, as individuals with a TSC2 variant presented with a higher frequency of cognitive impairment and ASD, but again the finding was not statistically significant. In the literature, multiple TAND symptoms are associated with approximately 90% of affected individuals. Reference Marcinkowska, Jóźwiak, Tarasewicz, Dębska-Ślizień and Szurowska17 In addition, it is important to consider that TAND symptoms may arise later in life due to the diversity of psychosocial factors that influence the diagnosis of neuropsychiatric symptoms. Reference Marcinkowska, Jóźwiak, Tarasewicz, Dębska-Ślizień and Szurowska17 Our cohort underrepresents this metric, which reinforces the idea that TAND symptoms may be undiagnosed or untreated when TSC is diagnosed later in life. Reference Marcinkowska, Jóźwiak, Tarasewicz, Dębska-Ślizień and Szurowska17 Studies report that TAND was only screened in 4% of patients at the time of their referral to a TSC clinic, Reference Alsowat, Zak and McCoy18 contributing to its underdiagnosis in adult patients. Our findings support the idea of reevaluation of TAND features on a regular basis. Reference Marcinkowska, Jóźwiak, Tarasewicz, Dębska-Ślizień and Szurowska17
As we did not have access to pediatric records, we cannot make confident conclusions on trends surrounding early age of onset of TSC symptoms, such as infantile spasms and epilepsy. Our patient health information regarding progression of epilepsy was inconsistent for those included in our analyses, thus we cannot make claims on the details of TSC-related epilepsy in adults. For the purpose of this study, we defined patients with epilepsy as those who presented with a history of multiple (two or more) seizures. Reference Stafstrom and Carmant19 Previous literature states that very little is known about the evolution of TSC-related epilepsy in adulthood. Reference Vignoli, La Briola and Turner20 However, our data revealed that a history of epilepsy was again more prevalent in patients with a variant in TSC2, which is also consistent with findings reported in previous studies. Reference Kothare, Singh and Chalifoux15 Our cohort presented with a rate of epilepsy of approximately 25% (12/51), compared to previous literature indicating 62%–93% of TSC patients will have epilepsy. Reference Nabbout, Belousova and Benedik21 Therefore, our data showed adults with TSC experienced epilepsy at lower rates and the burden of epilepsy may decrease over time. Moreover, about 70% of individuals with TSC experience seizure onset in the first year of life – making a de novo occurrence of epilepsy unlikely in adulthood, though little is known about the evolution of epilepsy in adulthood. Reference Vignoli, La Briola and Turner20 According to a study within a cohort of 231 patients, only four patients (12%) without a history of seizures developed epilepsy in adulthood, demonstrating an overall decreased risk with age. Reference Chu-Shore, Major, Camposano, Muzykewicz and Thiele22 However, some literature reports that epilepsy in adulthood may be underreported, especially in milder cases where clinical features may be more subtle or infrequent and this may have been the case in our cohort too. Reference Nabbout, Belousova and Benedik21
Patients with a variant of TSC2 also present with an increased rate of systemic involvement. Reference Bachour, House and Andrade23 Renal involvement, including both AMLs and cysts, was seen more frequently in those with a variant in TSC2 in our cohort. However, when compared with other genotypes, this finding was not statistically significant. Previous studies have reported similar trends. Reference Dabora, Jozwiak and Franz24 The TSC2 and PKD1 genes lie adjacent to each other on chromosome 16p, thus large deletions can disrupt the function of both genes. Reference Brook-Carter, Peral and Ward25 Of the two patients who had a whole deletion of TSC2/PKD1 genes, hepatic involvement was observed only in the TSC2 group, which is consistent with previous studies. Reference Dabora, Jozwiak and Franz24 Cardiac involvement between the two groups varied in the literature; Reference Hinton, Prakash, Romp, Krueger and Knilans26 our data showed that cardiac rhabdomyoma was observed in patients with TSC2 and NMI, but not TSC1 variants. Since we did not have access to pediatric records, trends regarding cardiac rhabdomyoma may be underestimated as they typically regress by adulthood. Reference Sciacca, Giacchi and Mattia27
Our findings regarding trends in adults with TSC were mostly consistent with those previously reported in the pediatric population, but our sample size was small, and the study was underpowered. Generally, patients in the TSC2 group showed increased systemic involvement and overall frequency of TSC-related features. Future directions include examining severity in relation to inheritance type (de novo, familial, mosaic) to better understand the molecular basis of TSC manifestations.
Some limitations to our study may include being a single-center cohort, therefore, limiting our data to a smaller set of patients. Even within our cohort, eight patients did not have genetic testing and were therefore not part of the overall analysis. Moreover, due to the nature of the clinical findings found within the various affected systems, some statistical analyses grouped more than one type of clinical manifestation. We also conducted multiple comparisons as part of our statistical analysis, which can increase the possibility of a type I error, and adjustments were not made to the P value in the study to account for this. Reference Lee and Lee28 In addition, the difference in TSC1 and TSC2 in the literature may be affected by reporting bias in different regions of the world. Reference Shlomovitz, Ben-Zeev and Pleniceanu29 The more severe phenotype in TSC2 deficient patients may result in a reporting bias, leading to an overrepresentation of TSC2 in the literature. Furthermore, we are also unable to look for somatic variants, as data regarding tumor testing were not obtained. Finally, the retrospective nature of our study brings a limited follow-up period and does not allow us to make claims on current experiences of patients involved in this study. Our study relied solely on retrospectively collected clinical data; therefore, it is possible that some findings are underreported. As data regarding age of diagnosis in our patient medical records were incomplete, we were unable to focus our analysis on the patients who were diagnosed with TSC in adulthood and further exploring age of onset for certain findings, such as epilepsy or cardiac rhabdomyoma.
Conclusions
Our study has explored and analyzed the large phenotypic variability associated with TSC genotypes in an adult population. Overall, our findings were consistent with previous literature, but elucidates lower epileptic trends amongst adults; which requires confirmation from other cohort studies. This supports that a multidisciplinary approach should be taken in order to manage TSC-related features in adults and provide appropriate care despite variants identified.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to acknowledge the following individuals from the Hospital for Sick Children (Toronto, Ontario) who developed the pediatric REDCap database: Mathieu Lemaire, Blathnaid McCoy and Maria Zak. Our appreciation to Derek Wong for his assistance in the statistical analysis, and Carolina Sanabria Salas for her assistance in the variant analysis.
Statement of authorship
This study was designed and supervised by RHK and KMF. Data curation and analysis were performed by DP with guidance from RW. Clinical care for the patients was provided by NF, MM and RHK. The first draft of the manuscript was written by DP and KMF, and all authors commented on previous versions of the manuscript. All authors reviewed and approved the final manuscript.
Funding statement
R.H.K. is supported by Anonymous (2), the Bhalwani Family Charitable Foundation, Goldie R. Feldman, Karen Green and George Fischer Genomics and Genetics Fund, Lindy Green Family Foundation, the FDC Foundation, the Shar Foundation, the Devine/Sucharda Charitable Foundation, the Princess Margaret Cancer Foundation, Leslie E. Born and the Hal Jackman Foundation. D.P. received support from the FDC Foundation.
Competing interests
None.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Research Ethics Committee at the University Health Network (#20-5813).
Consent to participate
A consent waiver was obtained by the University Health Network Research Ethics Board for this study.





 Open access
Open access