


Acute episodes of mania associated with bipolar disorder require immediate, rapid and cost-effective treatment; however, debate continues over the most appropriate first-line therapy. Options include monotherapy with a mood stabiliser or an antipsychotic drug or combination therapy with two mood stabilisers or a mood stabiliser plus an antipsychotic. Although combination therapy is commonly used in clinical practice and may offer an advantage over monotherapy (Reference Sachs, Printz and KahnSachs et al, 2000; American Psychiatric Association, 2002), few well-controlled studies have examined the efficacy of such an approach. Therefore, in this two-part, 13-week, phase III trial, we ascertained the efficacy and safety of risperidone add-on therapy to a mood stabiliser in the manic phase of bipolar disorder. We present the results from the first part, a randomised, 3-week, double-blind, placebo-controlled study. Results of the extension phase, an open-label study lasting up to 10 weeks, will be reported later.
METHOD
The study was conducted at sites in Canada, Israel, Norway, South Africa, Spain and the UK. The recruitment began on 2 October 1997 and the trial concluded on 6 October 1999. Prior to randomisation, prospective participants completed a screening period during which informed consent was obtained, eligibility was assessed and medical and psychiatric examinations were completed. The medical examination comprised electrocardiography (ECG), laboratory evaluations and measurement of the plasma concentration of the mood-stabilising agent. The psychiatric examination included assessment using the Young Mania Rating Scale (YMRS; Reference Young, Biggs and ZieglerYoung et al, 1978).
Inclusion and exclusion criteria
Eligible patients were 18-65 years old, had a DSM-IV bipolar disorder with a manic or mixed episode (American Psychiatric Association, 1994), with a minimum baseline score of 20 on the YMRS. Patients with concurrent symptoms of depression could be entered. Patients were included if they had been receiving a mood stabiliser — lithium, divalproex (sodium valproate plus valproic acid) or carbamazepine — for a minimum of 2 weeks prior to screening; in the event that the patient had not been receiving a mood stabiliser, one must have been prescribed prior to randomisation. Patients were medically stable, and were randomised within 7 days of hospital admission.
Patients were excluded if they had another DSM—IV Axis I diagnosis other than nicotine or caffeine dependence, a seizure disorder requiring medication, or a history of alcohol or drug misuse or dependence within the 3 months prior to the study. People at imminent risk of causing injury to themselves or others or of causing property damage were also excluded, as were people with serious or unstable medical disease, clinically significant laboratory abnormalities, severe drug allergy or hypersensitivity, or a history of neuroleptic malignant syndrome. Pregnant or nursing women and those of childbearing potential without adequate contraception also were excluded.
Patient population
Participants with acute mania who fulfilled the entry criteria were randomised to receive risperidone or placebo, with stratification for type of mood stabiliser (lithium, divalproex or carbamazepine), site, and whether mood stabiliser therapy had been initiated at the start of the trial or had been given for at least 2 weeks before the patient's screening visit, using a dynamic randomisation method. This method was based upon the minimisation technique (Reference White and FreedmanWhite & Freedman, 1978) and was implemented by each site telephoning the coordinating centre to obtain the randomisation number for each patient; it ensured balanced treatment groups on the factors of mood stabiliser and time of initiation of mood stabiliser therapy. Patients underwent a ‘wash-out’ period of 3 days, unless the investigator believed that antipsychotic medication was needed sooner. During the wash-out period antipsychotic, anti-Parkinsonian, anti-depressant, anxiolytic and other centrally acting drugs were discontinued. Flurazepam, temazepam, oxazepam and chloral hydrate were allowed as sleep aids. After initiation of double-blind treatment, patients remained in hospital for at least 4 days. Study visits were scheduled at screening, at baseline (day 1) and at days 3, 8, 15 and 22.
Patients were withdrawn from the study if they retracted consent, violated the randomisation code, discontinued mood stabiliser therapy for more than 4 consecutive days, or had a serious adverse event. After completion of the 3-week double-blind study — or after completing at least 7 days of double-blind treatment — participants were eligible to enter a 10-week, open-label extension study. The double-blind code was not broken, but every participant who entered the second phase of the study received open-label risperidone.
Study medication
All study participants were initially treated with 2 mg risperidone or placebo (tablets) once daily on days 1 and 2. On days 3 and 4 the risperidone dose could be increased to a maximum of 4 mg daily or decreased to 1 mg daily. On days 5 to 21 the dose could be increased to a maximum of 6 mg daily or decreased to a minimum of 1 mg. Venous blood samples were taken at baseline and on day 22 to determine plasma concentrations of risperidone and its active metabolite 9-hydroxyrisperidone, using a radioimmunoassay.
During the double-blind period of treatment all patients also received lithium, divalproex or carbamazepine. Only one mood stabiliser at a time was permitted; another drug could be substituted only for safety reasons, not for efficacy. The dosage of the mood stabiliser was reduced whenever an adverse event attributable to the drug occurred. If the adverse effect persisted, another mood stabiliser could be substituted with no more than a 2-day overlap. Patients whose mood stabiliser was switched retained their original prescription stratification for purposes of statistical analysis. Plasma drug concentrations were measured whenever clinically indicated and at screening, baseline and on days 3, 8, 15 and 22, and the doses of the mood stabilisers were adjusted to achieve therapeutic levels.
Prior and concomitant therapy
Medications not allowed during the study included antipsychotic agents other than the trial medication; mood stabilisers other than lithium, divalproex or carbamazepine; and benzodiazepines other than lorazepam, oxazepam, temazepam or flurazepam. Patients were permitted to take up to 6 mg lorazepam daily for agitation during the wash-out period and up to 4 mg daily during the first 7 days of the double-blind period. Anti-Parkinsonian medication was not permitted at baseline, but could be prescribed for extrapyramidal symptoms after administration of the Extrapyramidal Symptom Rating Scale (Reference Chouinard, Ross-Chouinard and AnnableChouinard et al, 1980). Antidepressant drugs were not permitted at the start of double-blind treatment but could be prescribed if clinically significant depression emerged, identified using the Hamilton Rating Scale for Depression (HRSD; Reference HamiltonHamilton, 1967), and if the investigator believed that such treatment was unlikely to worsen manic symptoms. Medication for ongoing medical conditions was continued.
Efficacy evaluations
Assessments using the YMRS (Reference Young, Biggs and ZieglerYoung et al, 1978), the Clinical Global Impression (CGI) scale (Reference GuyGuy, 1976), the Brief Psychiatric Rating Scale (BPRS; Reference Overall and GorhamOverall & Gorham, 1962) and a 21-item HRSD evaluation were completed at baseline and on days 8, 15 and 22.
The primary measure of efficacy was the change in the YMRS score from baseline to end-point, which was the last available observation for each patient. Other parameters included the YMRS score change from baseline to day 8, the percentage of patients showing a 50% or greater improvement on the YMRS, and the time to onset of therapeutic response (in days) as represented by a reduction of at least 30% in the YMRS score. Further measures of efficacy included the changes from baseline in CGI, BPRS and HRSD scores, and the percentage of patients who used adjunctive lorazepam.
Safety evaluations
Vital signs were measured at screening, at baseline, and on days 8, 15 and 22. A physical examination including weight, ECG and laboratory evaluations was performed at screening and on day 22. The Extrapyramidal Symptom Rating Scale (ESRS), administered at baseline and on days 8, 15 and 22, consisted of a questionnaire; parkinsonism, dystonia and dyskinesia sub-scales; a clinical global impression of overall severity of dystonia, parkinsonism and dyskinesia; and parkinsonism staging.
Statistical analysis
Assuming a standard deviation of 12.2, a total of 132 participants was required to detect a six-point difference between groups in the mean YMRS score change from baseline (0.05 significance, two-tailed, with 80% power). To adjust for drop-outs, the total number of randomised participants was increased to 150 (75 per group). No power calculation has been performed for any of the secondary efficacy measures, nor have the P values been adjusted for multiplicity.
For the change from baseline to end-point in YMRS score, an analysis of covariance model was used to test for differences between treatment groups including factors for treatment, country, strata (initiation of mood stabiliser therapy and type of drug), baseline score (as covariate), and their interactions with treatment. A similar analysis was used for changes from baseline in the BPRS and HRSD scores. Clinical Global Impression results are based on the van Elteren test, which evaluates the overall difference between treatments based on linear combinations of Wilcoxon statistics (Reference van Elterenvan Elteren, 1960). For measures without a baseline response (e.g. percentage of days on which patients used adjunctive lorazepam) an analysis of variance (ANOVA) model was used, which contained treatment, country and strata factors.
The Cochran—Mantel—Haenszel test for general association, controlling for country, was used to test for treatment differences in the clinical response rates on the YMRS and in the percentage of patients who used adjunctive lorazepam. All statistical tests were interpreted at the 0.05 significance level (two-tailed).
RESULTS
Patient demographics and disposition
A total of 157 participants entered the screening phase: of these, 151 were randomised to treatment, 75 to risperidone plus mood stabiliser and 76 to placebo plus mood stabiliser. One patient randomised to the placebo group withdrew consent before study medication was administered. Both groups had similar baseline characteristics. Approximately 10% of the patients in each group had experienced a mixed episode. Thirty-eight patients (51%) assigned to the risperidone group and 47 (63%) of the placebo group were free of psychotic features at baseline (Table 1). Sixty-nine patients randomised to the risperidone group and 73 of those receiving placebo had at least two assessments and were included in the efficacy analysis. A total of 48 patients (64%) in the risperidone group and 36 (48%) in the placebo group completed the 3-week double-blind phase (mean difference in completion rates 16%; 95% CI 0.32-31.68) (Table 2, Fig. 1).

Fig. 1 Study profile. DB, double-blind study phase; OL, open-label study phase.
Table 1 Demographic data and baseline disease characteristics

| Risperidone+mood stabiliser (n=75) | Placebo+mood stabiliser (n=75)1 | |
|---|---|---|
| Gender (n (%)) | ||
| Male | 32 (43) | 31 (41) |
| Female | 43 (57) | 44 (59) |
| Age in years (median (range)) | 37 (20-63) | 42 (19-65) |
| Axis I diagnosis (n (%)) | ||
| Bipolar disorder, manic | 70 (93) | 68 (91) |
| Bipolar disorder, mixed | 5 (7) | 7 (9) |
| Current episode (n (%)) | ||
| Mild severity | 3 (4) | 2 (3) |
| Moderate severity | 21 (28) | 28 (37) |
| Severe with psychotic features | 37 (49) | 28 (37) |
| Severe without psychotic features | 14 (19) | 17 (23) |
Table 2 Patient disposition: reasons for discontinuations
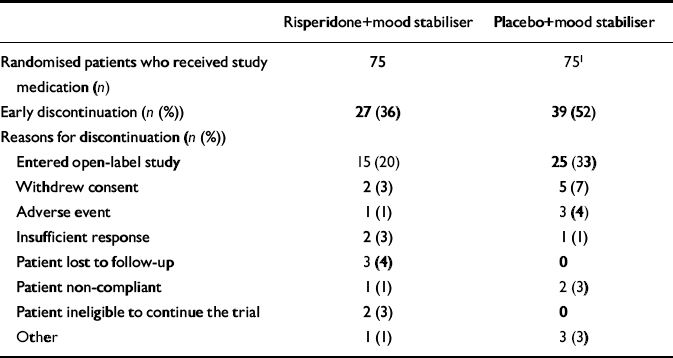
| Risperidone+mood stabiliser | Placebo+mood stabiliser | |
|---|---|---|
| Randomised patients who received study medication (n) | 75 | 751 |
| Early discontinuation (n (%)) | 27 (36) | 39 (52) |
| Reasons for discontinuation (n (%)) | ||
| Entered open-label study | 15 (20) | 25 (33) |
| Withdrew consent | 2 (3) | 5 (7) |
| Adverse event | 1 (1) | 3 (4) |
| Insufficient response | 2 (3) | 1 (1) |
| Patient lost to follow-up | 3 (4) | 0 |
| Patient non-compliant | 1 (1) | 2 (3) |
| Patient ineligible to continue the trial | 2 (3) | 0 |
| Other | 1 (1) | 3 (3) |
Medications
For each patient assigned to the risperidone group the modal daily dose of risperidone was calculated (i.e. the most frequently used risperidone dose throughout the treatment period). The median modal dose of risperidone was 4.0 mg. The treatment duration was as follows: for the placebo group, median 18 days (quartiles 7, 21, 28); for the risperidone group, median 21 days (quartiles 12, 21, 24). The median plasma concentration of the active moiety (sum of risperidone and 9-hydroxyrisperidone) at end-point was 17.2 μg/l (first and third quartiles 0.0, 36) and the median dose normalised (dose corrected with the 4 mg dose as a reference point, making it possible to interpret certain effects by excluding the dose as a confounding factor) concentration was 14.6 μg/l (quartiles 0.0, 25.9). The plasma concentrations of risperidone, 9-hydroxyrisperidone and the active moiety were similar when lithium or divalproex were taken concurrently; the median dose-normalised concentrations for the active moiety were 17.1 μg/l (quartiles 0.0, 40.6) or 23.4 μg/l (quartiles 0.0, 38.1), respectively. When risperidone was co-administered with carbamazepine, however, median dose-normalised plasma concentrations of the active moiety were approximately 40% lower (10 μg/l quartiles 5, 21.6) at the end of the 3-week double-blind phase.
Fewer than half of the patients (43%) had been receiving a mood stabiliser before entering the trial. At baseline, 86 (57%) of the patients received lithium, 38 (25%) received divalproex and 26 (17%) received carbamazepine (Table 3). At weeks 1 to 3, plasma concentrations of mood-stabilising agents were within the targeted therapeutic range for all groups. Seven patients (five in the risperidone group, two in the placebo group) switched to a different mood stabiliser or, in the switching process, overlapped two different mood stabilisers during the study.
Table 3 Use of mood stabilisers

| Risperidone group (n=75) | Placebo group (n=75)1 | |
|---|---|---|
| Initiation of mood stabiliser treatment (n (%)) | ||
| Prior to the study | 30 (40) | 35 (47) |
| At the start of the study | 45 (60) | 40 (53) |
| Patients who received a mood stabiliser at baseline (n (%)) | ||
| Lithium | 42 (56) | 44 (59) |
| Divalproex | 19 (25) | 19 (25) |
| Carbamazepine | 14 (19) | 12 (16) |
Adjunctive lorazepam
Fifty-four patients (72%) in the risperidone group and 47 (63%) in the placebo group used lorazepam during the first 7 days (mean difference 9%; 95% CI - 5.9 to 23.9). The mean percentage of days that lorazepam was used was 44% in the risperidone group and 58% in the placebo group (P=0.02; between-group difference 13.5, 95% CI - 25.0 to - 1.9).
Efficacy based on YMRS
Baseline YMRS scores were similar in the two treatment groups (Table 4). At week 1, the risperidone group showed significantly greater improvement as indicated by decreases in YMRS scores relative to baseline (- 10.2) compared with the placebo group (- 6.7; 95% CI - 6.35 to - 0.35). On the efficacy measure of mean change in YMRS scores from baseline to end-point, the risperidone group had a mean decrease of 14.5 points (49%) in YMRS scores while the placebo group had a mean decrease of 10.3 points (36%) in YMRS scores (mean difference in change score - 4.2; 95% CI - 7.60 to 0.54) (Table 4). At end-point, 40 patients (59%) in the risperidone group responded (defined as 50% or greater reduction in YMRS scores from baseline) compared with 30 (41%) in the placebo group (mean difference 17.7%, 95% CI 0.8-33.5; P < 0.05).
Table 4 Total mean scores on the Young Mania Rating Scale (YMRS) at baseline and mean change from baseline during double-blind treatment
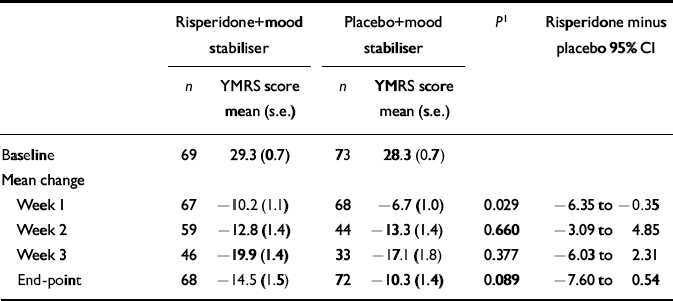
| Risperidone+mood stabiliser | Placebo+mood stabiliser | P 1 | Risperidone minus placebo 95% CI | |||
|---|---|---|---|---|---|---|
| n | YMRS score mean (s.e.) | n | YMRS score mean (s.e.) | |||
| Baseline | 69 | 29.3 (0.7) | 73 | 28.3 (0.7) | ||
| Mean change | ||||||
| Week 1 | 67 | -10.2 (1.1) | 68 | -6.7 (1.0) | 0.029 | -6.35 to -0.35 |
| Week 2 | 59 | -12.8 (1.4) | 44 | -13.3 (1.4) | 0.660 | -3.09 to 4.85 |
| Week 3 | 46 | -19.9 (1.4) | 33 | -17.1 (1.8) | 0.377 | -6.03 to 2.31 |
| End-point | 68 | -14.5 (1.5) | 72 | -10.3 (1.4) | 0.089 | -7.60 to 0.54 |
Participants found to have psychotic features at baseline who received risperidone had a mean reduction in YMRS score of 15.1 from baseline to end-point, while those randomised to placebo had a mean reduction of 12.2. Among participants without psychotic features, those in the risperidone group had a mean reduction in YMRS score of 13.8 while those in the placebo group had a mean reduction of 9.2. Analysis of covariance showed no main effect of psychotic features on YMRS score change.
Patients who began taking a mood stabiliser at the start of the study and were randomised to risperidone (n=42) had a mean reduction in YMRS score of 14.9 from baseline to end-point, while patients randomised to placebo (n=39) had a mean reduction of 13.2 (mean difference 1.55; 95% CI - 3.78 to 6.87). In patients who had been taking a mood stabiliser for at least 2 weeks prior to screening, the mean reduction in YMRS score from baseline to end-point was 13.8 in the risperidone group (n=26) and 7.1 in the placebo group (n=34) (mean difference 6.30; 95% CI - 0.008 to 12.61).
Further measures of efficacy
Clinical Global Impression
The CGI severity ratings at baseline were comparable in both groups, with most patients having marked or moderate manic symptoms. Both at the end of the first week of treatment and at end-point, the distributions of entire CGI improvement scores of the risperidone group were more concentrated on the ‘very much improved’ and ‘much improved’ categories compared with the placebo group (P=0.013 at week 1 and P=0.022 at end-point using the van Elteren test to control for country). For example, 48% (n=31) at week 1 and 61% (n=40) at end-point of the risperidone group had ‘much’ or ‘very much’ improvement on the CGI scale compared with 31% (n=21) at week 1 and 43% (n=31) at end-point in the placebo group (mean difference in responders at week 1 16.8%, 95% CI 0.7-32.9; mean difference in responders at end-point 17.5%, 95% CI 1.1-33.9).
Brief Psychiatric Rating Scale
Both treatment groups had comparable baseline BPRS total and sub-scale scores. Patients assigned to receive risperidone had significantly greater improvement on total BPRS scores at week 1 and at end-point compared with the placebo group (Table 5). Furthermore, at end-point the risperidone group had significantly greater improvement in the hostility and thought disturbance sub-scales of the BPRS than did the placebo group (P < 0.05). In the analysis of activity and anxiety/depression sub-scales, improvement with risperidone tended to be greater than that with placebo.
Table 5 Scores on the Brief Psychiatric Rating Scale (BPRS) at baseline and mean change from baseline
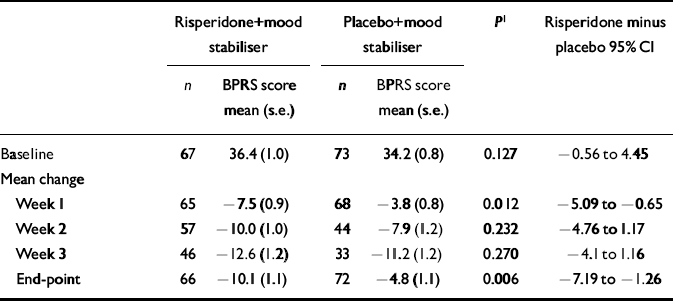
| Risperidone+mood stabiliser | Placebo+mood stabiliser | P 1 | Risperidone minus placebo 95% CI | |||
|---|---|---|---|---|---|---|
| n | BPRS score mean (s.e.) | n | BPRS score mean (s.e.) | |||
| Baseline | 67 | 36.4 (1.0) | 73 | 34.2 (0.8) | 0.127 | -0.56 to 4.45 |
| Mean change | ||||||
| Week 1 | 65 | -7.5 (0.9) | 68 | -3.8 (0.8) | 0.012 | -5.09 to -0.65 |
| Week 2 | 57 | -10.0 (1.0) | 44 | -7.9 (1.2) | 0.232 | -4.76 to 1.17 |
| Week 3 | 46 | -12.6 (1.2) | 33 | -11.2 (1.2) | 0.270 | -4.1 to 1.16 |
| End-point | 66 | -10.1 (1.1) | 72 | -4.8 (1.1) | 0.006 | -7.19 to -1.26 |
Hamilton Rating Scale for Depression
At baseline, the mean HRSD scores for the two groups were comparable (risperidone 8.6, placebo 8.2). No significant difference was present between the two groups in changes from baseline in total and cluster HRSD scores at weeks 1, 2 and 3 or at end-point. At end-point the mean decreases in HRSD total scores were 4.1 for the risperidone group and 2.1 for the placebo group. Two patients in the placebo group and one patient in the risperidone group experienced depressive symptoms during the study and were prescribed antidepressants.
Post hoc analysis
The markedly lower plasma concentrations of the active moiety of risperidone seen in patients who received carbamazepine prompted a post hoc analysis of the YMRS change scores from baseline to end-point in patients who received lithium or divalproex. In this analysis, which excluded patients who received carbamazepine, the YMRS change scores of the risperidone group were significantly greater than those of the placebo group at end-point (P=0.047) and at week 1 (P=0.038) (Table 6).
Table 6 Mean scores on the Young Mania Rating Scale (YMRS), excluding the carbamazepine subgroup
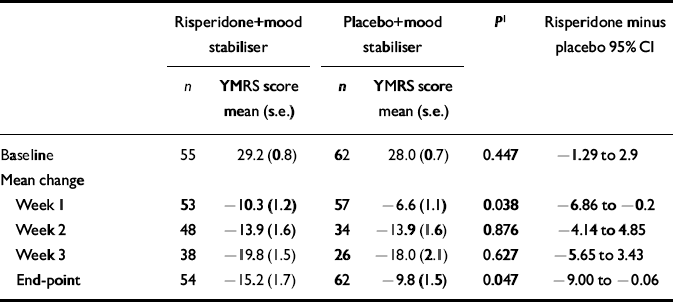
| Risperidone+mood stabiliser | Placebo+mood stabiliser | P 1 | Risperidone minus placebo 95% CI | |||
|---|---|---|---|---|---|---|
| n | YMRS score mean (s.e.) | n | YMRS score mean (s.e.) | |||
| Baseline | 55 | 29.2 (0.8) | 62 | 28.0 (0.7) | 0.447 | -1.29 to 2.9 |
| Mean change | ||||||
| Week 1 | 53 | -10.3 (1.2) | 57 | -6.6 (1.1) | 0.038 | -6.86 to -0.2 |
| Week 2 | 48 | -13.9 (1.6) | 34 | -13.9 (1.6) | 0.876 | -4.14 to 4.85 |
| Week 3 | 38 | -19.8 (1.5) | 26 | -18.0 (2.1) | 0.627 | -5.65 to 3.43 |
| End-point | 54 | -15.2 (1.7) | 62 | -9.8 (1.5) | 0.047 | -9.00 to -0.06 |
Trial discontinuations
Twelve patients (16%) in the risperidone group and 14 (19%) in the placebo group discontinued treatment early (difference in proportion discontinuing early -3%; 95% CI -15.1 to 9.1). Another 15 patients (20%) in the risperidone group and 25 patients (33%) in the placebo group left the 3-week double-blind phase of the study early and entered the open-label extension phase (difference in proportion -13%; 95% CI -26.9 to 0.9).
Safety
The incidence of adverse events was similar in the two groups: 57% of the risperidone group and 51% of the placebo group reported at least one adverse event (between-group difference in overall adverse event rate 6%; 95% CI -9.9 to 21.9). The most frequently reported adverse events, excluding extrapyramidal-related adverse events, were headache (9% in the risperidone group, 9% in the placebo group), insomnia (4% and 8%) and nausea (5% and 3%). One patient in each group had worsening of manic symptoms. Extrapyramidal-related adverse events were reported by 16 patients in the risperidone group and 6 patients in the placebo group (P=0.013, Cochran—Mantel—Haenszel test for general association controlling for country, Table 7). Each group had similar ESRS total scores at baseline and end-point (both groups had a mean change from baseline of -0.1). Twelve patients in the risperidone group and 6 in the placebo group used anti-Parkinsonian medication (P=0.108, Cochran—Mantel—Haenszel test for general association controlling for country). Among patients who received anti-Parkinsonian medication, those in the risperidone group used it for 49% of the study days; those in the placebo group used it for 64% of the study days (P=0.236, ANOVA model with factors for treatment, stratification and country).
Table 7 Extrapyramidal symptom-related adverse events
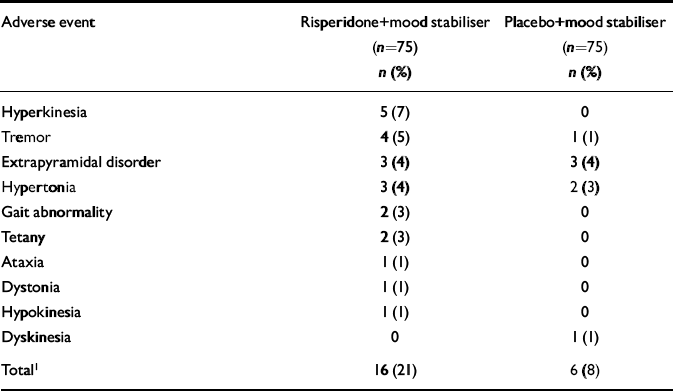
| Adverse event | Risperidone+mood stabiliser (n=75) n (%) | Placebo+mood stabiliser (n=75) n (%) |
|---|---|---|
| Hyperkinesia | 5 (7) | 0 |
| Tremor | 4 (5) | 1 (1) |
| Extrapyramidal disorder | 3 (4) | 3 (4) |
| Hypertonia | 3 (4) | 2 (3) |
| Gait abnormality | 2 (3) | 0 |
| Tetany | 2 (3) | 0 |
| Ataxia | 1 (1) | 0 |
| Dystonia | 1 (1) | 0 |
| Hypokinesia | 1 (1) | 0 |
| Dyskinesia | 0 | 1 (1) |
| Total1 | 16 (21) | 6 (8) |
No clinically significant change in vital signs or laboratory values was observed in either group. At baseline, the mean body weight of the risperidone group was 76.5 kg and that of the placebo group was 74.3 kg. At end-point, the mean weight increase in the former group was 1.7 kg and in the latter 0.5 kg (P=0.012, ANOVA model with factors for treatment, stratification and country). Small fluctuations in the mean value of ECG parameters were observed during the course of the trial, none of which was considered clinically relevant. No significant between-group difference in ECG changes from baseline was observed.
DISCUSSION
Efficacy on YMRS
The results of this study indicate that risperidone, at a median modal dose of 4 mg, is more efficacious than placebo when combined with mood-stabilising therapy in the treatment of manic episodes associated with bipolar disorder. Improvement in manic symptoms with risperidone combination therapy was rapid, as indicated by significantly greater reductions in YMRS scores from baseline to week 1. At end-point, a significantly greater number of patients met criteria for response on the YMRS in the risperidone group compared with those in the placebo group. These results are consistent with those reported in two small, uncontrolled studies (Reference Ghaemi, Sachs and BaldassanoGhaemi et al, 1997; Reference Ghaemi and SachsGhaemi & Sachs, 1997) and in a large open-label study (Reference Vieta, Corbella and ReinaresVieta et al, 2001), and support the findings of a double-blind, randomised, controlled trial that compared risperidone monotherapy with haloperidol and lithium monotherapies (Reference Segal, Berk and BrookSegal et al, 1998).
Further measures of efficacy
Risperidone in combination with mood-stabilising therapy was associated with more rapid and significantly greater improvements in the BPRS and CGI measures compared with placebo plus mood-stabilising therapy. More patients in the risperidone group had much or very much improvement on the CGI improvement scale compared with those in the placebo group. Furthermore, hostility, thought disturbance and activity were better controlled with risperidone than with placebo in combination with mood-stabilising therapy. This was further supported by the observation that patients in the placebo group required adjunctive lorazepam for a greater number of days than those in the risperidone group. Thus, risperidone plus mood-stabilising treatment may reduce the overall burden, staff time and other costs associated with acute mania, particularly as many patients with bipolar disorder require long-term treatment.
Risperidone plus a mood stabiliser was equally effective in patients with or without psychotic features, as indicated by similar magnitude of reductions in YMRS scores in both groups. This finding, supported by Ghaemi and colleagues (Reference Ghaemi, Sachs and BaldassanoGhaemi et al, 1997), suggests that risperidone — like other atypical antipsychotic agents such as olanzapine (Reference Tohen, Jacobs and GrundyTohen et al, 2000) — has antimanic properties independent of its antipsychotic properties. In addition, improvement in measures of anxiety and depression in this study tended to be greater in patients who received risperidone rather than placebo in combination with a mood stabiliser.
Effect of carbamazepine on risperidone plasma levels and efficacy
Reductions in YMRS score from baseline to end-point tended to be greater in the risperidone group (14.5) than in the placebo group (10.3). The magnitude of difference in YMRS change scores between the risperidone and placebo groups (4.2) observed in this study was comparable to that reported between olanzapine add-on and placebo add-on groups (4.01) in a similar study of 6 weeks' duration (Reference Tohen, Chengappa and SuppesTohen et al, 2002). Furthermore, the magnitude of reduction in YMRS scores in the risperidone group in this study might have been blunted by the conspicuous effect of carbamazepine on risperidone plasma concentrations. This possibility is supported by the observation that plasma levels of risperidone active moiety were approximately 40% lower in the carbamazepine group compared with those in the lithium or divalproex group and by the results of post hoc analysis showing significant reductions in YMRS score from baseline to end-point after exclusion of the carbamazepine group. Because risperidone doses were not adjusted upwards in the carbamazepine group within the limited period of this trial, optimal concentrations might not have been achieved in that group. Based on our results and on those of others (Reference Freeman and StollFreeman & Stoll, 1998), patients who receive risperidone or other psychotropic agents concomitantly with carbamazepine should be monitored closely, and dosages should be adjusted if necessary.
Effect of risperidone on mania
Several case reports suggest an association between risperidone treatment and hypomanic or manic episodes in patients with bipolar or schizoaffective disorder (Reference Aubrey, Simon and BertskyAubrey et al, 2000). Patients who experienced these effects generally received high doses of risperidone and concomitant mood stabilisers were abruptly discontinued. The results of this placebo-controlled, double-blind trial indicate that risperidone does not worsen mania. This is consistent with results of a large, open-label study which showed that risperidone is not associated with the induction of mania (Reference Vieta, Corbella and ReinaresVieta et al, 2001).
Adverse events
The risperidone and placebo combinations with a mood stabiliser were equally well tolerated. Although patients in the risperidone group reported extrapyramidal-related adverse events more frequently, the ESRS change scores were similar in both groups suggesting that risperidone therapy at the dosages used in this study was not associated with significant extrapyramidal symptoms. This was supported by the absence of any significant difference in the use of anti-Parkinsonian medication between the two groups.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Risperidone prescribed in addition to a mood stabiliser can more rapidly (1 week) improve acute manic episodes than a mood stabiliser alone; more patients showed improvement in mania compared with those on mood-stabilising therapy alone.
-
▪ Risperidone was efficacious in patients both with and without psychotic features.
-
▪ Risperidone did not worsen mania or induce depression.
LIMITATIONS
-
▪ The primary efficacy measure (change in score on the Young Mania Rating Scale from baseline to end-point) was only marginally statistically significant.
-
▪ The post hoc analysis of the primary efficacy measure was not based on an a priori hypothesis.
-
▪ Many patients dropped out of the 3-week double-blind phase.









eLetters
No eLetters have been published for this article.