


Bipolar disorder is a lifelong episodic illness requiring long-term treatment, yet there remains a need for well-tolerated and clinically effective maintenance of effect and continuation therapies. Aripiprazole has a novel pharmacological profile; partial agonism at dopamine D2 and D3 and serotonin 5-HT1A receptors, and antagonism at 5-HT2A receptors. Reference Burris, Molski, Xu, Ryan, Tottori and Kikuchi1–Reference Jordan, Koprivica, Dunn, Tottori, Kikuchi and Altar5 Aripiprazole has been shown to improve symptoms of acute mania in placebo-controlled studies Reference Keck, Marcus, Tourkodimitris, Ali, Liebeskind and Saha6,Reference Sachs, Sanchez, Marcus, Stock, McQuade and Carson7 and was superior to placebo in maintaining efficacy for up to 100 weeks in patients with bipolar mania. Reference Keck, Calabrese, McQuade, Carson, Carlson and Rollin8,Reference Keck, Calabrese, McIntyre, McQuade, Carson and Eudicone9 This study used a randomised placebo-controlled study design and included an active haloperidol reference arm to evaluate the acute and continued efficacy of aripiprazole monotherapy in patients with bipolar disorder experiencing acute manic or mixed episodes (trial registration NCT00097266).
Methods
Patients
Eligible patients were aged 18 years or older with bipolar I disorder manic or mixed type (with or without psychotic features), as defined by DSM–IV 10 and confirmed by the Mini International Neuropsychiatric Interview (MINI), who were experiencing an acute relapse requiring hospitalisation. In addition, they had a screening and baseline Young Mania Rating Scale (YMRS) Reference Young, Biggs, Ziegler and Meyer11 Total score ≥20, with less than a 25% decrease between the two visits. Patients also had a baseline Montgomery–Åsberg Depression Rating Scale (MADRS) Reference Montgomery and Åsberg12 Total score ≤17, with no more than a 4-point increase between the two visits. Female patients of child-bearing potential were required to use acceptable contraceptive measures.
Patients were excluded if they had delirium, dementia, amnestic or other cognitive disorders, schizophrenia or schizoaffective disorder, or if they were experiencing their first manic or mixed episode. Also excluded were patients with borderline, paranoid, histrionic, schizotypal, schizoid or antisocial personality disorder.
Other exclusions were: serious, unstable medical illness; hospitalisation for current mania or mixed episode for >3 weeks; previously unresponsive to treatments for manic symptoms (based on clinical judgement that the patient failed treatment with appropriate antimanic therapies (antipsychotic or mood stabiliser such as lithium, valproate, carbamazepine or haloperidol) at a clinically appropriate dose and duration); diagnosis of bipolar II disorder; rapid cycling bipolar disorder (>4 manic or depressive episodes/year); DSM–IV-defined substance misuse or substance dependence; clinically assessed significant risk of suicide; recent treatment with long-acting antipsychotics; use of mood stabilisers or antidepressants 2–4 weeks prior to randomisation; and electro-convulsive therapy within the past 3 months.
The study was conducted at 59 study centres in Bulgaria, Croatia, Mexico, Peru, Russia, South Africa and the USA. All patients provided written informed consent before participation. The study protocol, procedures and consent statement were approved by the study sites' institutional review boards.
Study design
This randomised double-blind placebo-controlled parallel-group study had three treatment arms. Following a wash-out period of 2–14 days, eligible patients were randomised to placebo (placebo group), aripiprazole (aripiprazole group) or haloperidol (haloperidol group) in a ratio of 1:1:1. Patients started aripiprazole at 15 mg/day or haloperidol 5 mg/day. At day 4, investigators could increase the daily dose (aripiprazole 30 mg/day; haloperidol 10 mg/day) as clinically indicated. The dose of haloperidol could be increased again at day 7 to 15 mg/day. Doses could be adjusted throughout the study based on tolerability and clinical response. Patients in the aripiprazole and haloperidol groups remained on treatment for 12 weeks, whereas patients on placebo were switched in a masked manner to double-blind aripiprazole at week 3. The masked aripiprazole dose depended on the number of placebo tablets administered at the end of week 3. These patients were not included in subsequent treatment comparisons.
For the first 2 weeks of treatment, patients remained hospitalised. At the end of week 2, patients could be discharged if they achieved ≤3 (mildly ill, minimally ill, not ill) on the Clinical Global Impressions – Bipolar version (CGI–BP) Reference Spearing, Port, Leverich, Brandt and Nolen13 Severity of Illness (Mania) score and ≤2 (much improved, very much improved) on the CGI–BP Severity of Illness Change from Preceding Phase (Mania) score. Patients who did not meet these criteria remained hospitalised. Patients who could not be discharged from hospital at the end of week 3 were discontinued from the study.
Concomitant medications
The following medications were prohibited during the study: antipsychotics, antidepressants, mood stabilisers, anticonvulsants, sleep aids, and all other psychotropic drugs (except benzodiazepines). Although benzodiazepines were permitted, the dose was tapered from ≤4 mg/day at days 1–4 to 0 mg/day at day 15. In addition, benzodiazepines were not permitted within 8 h prior to clinic assessments. Anticholinergic therapy (benztropine 4 mg/day, biperiden or trihexyphenidyl) was permitted for the treatment of extrapyramidal symptoms. Propranolol was permitted at a maximum dose of 60 mg/day for the treatment of akathisia or tremor.
Efficacy assessments
Treatment efficacy was assessed at baseline and at days 2, 4, 7 and 10 and thereafter at the end of weeks 2, 3, 4, 5, 6, 8, 10 and 12. The primary a priori efficacy outcome measure was mean change from baseline to week 3 in the YMRS Total score (last observation carried forward (LOCF)). Changes in YMRS Total scores from baseline were also analysed using a mixed-model repeated measures (MMRM) analysis on the observed case data-set to confirm the findings of LOCF. The key secondary end-point was mean change from baseline in the CGI–BP Severity of Illness (Mania) score. Other secondary efficacy end-points included response rate (proportion of patients demonstrating a ≥50% improvement in YMRS Total score), mean CGI–BP Severity of Illness Change from Preceding Phase (Mania) score, remission rate (proportion of patients achieving a YMRS Total score ≤12), mean change from baseline in YMRS Total score at other time points, MADRS Total score, Positive and Negative Syndrome Scale (PANSS) Total scores and Cognitive, Hostility, Positive and Negative sub-scale scores, Reference Kay, Fiszbein and Opler14,Reference White, Harvey, Opler and Lindenmayer15 and Longitudinal Interval Follow-up Evaluation – Range of Impaired Function Tool (LIFE–RIFT) Reference Leon, Solomon, Mueller, Endicott, Posternak and Judd16 Total score. In addition, the mean changes from baseline in YMRS Total score and CGI–BP Severity of Illness (Mania) score for all patients who were responders at week 3 were derived for each study visit.
Safety and tolerability assessments
Vital sign measurements were made initially at randomisation, and then on a weekly basis until week 3 and subsequently at weeks 8 and 12. Routine laboratory tests and weight measurements were carried out at screening, baseline, week 3 and week 12. Adverse event reports were recorded at each assessment. Extrapyramidal symptoms were evaluated using the Simpson–Angus Scale (SAS), Reference Simpson and Angus17 the Barnes Akathisia Rating Scale (BARS) Reference Barnes18 and the Abnormal Involuntary Movement Scale (AIMS). Reference Guy19
Statistical procedures
A total of 156 patients were estimated per treatment group to give 95% power to detect a 5.5-point difference in mean change in YMRS Total score at week 3 between aripiprazole and placebo at an α-level of 0.05. In addition, a sample size of 156 per treatment group was estimated to give 90% power to detect a 5.5-point difference in the mean change in YMRS Total score at week 3 between haloperidol and placebo using a hierarchical testing procedure to preserve the significance level at 0.05.
Efficacy parameters were analysed on an intent-to-treat basis using both LOCF and observed case data-sets. Continuous efficacy measures were evaluated on the LOCF data-set using analysis of covariance (ANCOVA) adjusted for baseline value, study centre and treatment. A hierarchical testing procedure was used to preserve the significance level at 0.05. Mean change from baseline to week 3 in YMRS Total score was first compared between aripiprazole and placebo using α=0.05. If aripiprazole was statistically significantly different from placebo, haloperidol was then compared with placebo at α=0.05. If the differences between aripiprazole and placebo and between haloperidol and placebo on the primary efficacy measure were statistically significant (P≤0.05), then testing of the difference between aripiprazole and placebo in the key secondary efficacy measure (mean change from baseline to week 3 (LOCF) in CGI–BP Severity of Illness (mania) score), could proceed at α=0.05. Testing of all the other secondary end-points was performed at the α=0.05 significance level without adjustment for multiple comparisons and multiple testing. Response and remission rates were analysed using the Cochran–Mantel–Haenszel general association statistic. Mixed-model repeated measures analysis was performed on change from baseline in YMRS Total score using direct likelihood estimation. The model included terms for baseline, visit, treatment, study centre, baseline × visit and treatment × visit. An unstructured covariance matrix was used to model within-patient error. Estimates of mean changes from baseline in metabolic parameters were obtained from ANCOVA adjusted for baseline and treatment. Post hoc week 12 comparisons between active treatments for change in YMRS Total scores, response and remission rates were carried out, although the study was not powered to find statistically significant differences at week 12.
Results
Patients
A total of 614 patients were enrolled in this study, of whom 485 were randomised to placebo (n=153), haloperidol (n=165) or aripiprazole (n=167). The proportion of patients completing each phase of the study was similar in each treatment arm (Fig. 1). At week 3, completion rates were similar for all treatment groups (aripiprazole 75%; haloperidol 73%; placebo 71%) and reasons for discontinuation were similar between groups. Week 12 completion rates were 57% for the aripiprazole group and 58% for the haloperidol group.

Fig. 1 Flow of participants through the study.
Baseline demographics and patient characteristics (online Table DS1) were similar between groups. At baseline, the mean body mass index (BMI) was 27.5 kg/m2; 43% of patients had a BMI >27 kg/m2.
Study treatments
The mean weekly mean dose of aripiprazole was 17.8 mg/day (n=166) for the initial 7 days of treatment, increasing to 23.6 mg/day at week 3 (n=144). At week 12, the mean weekly mean dose was 22.0 mg/day (n=96). The mean weekly mean dose of haloperidol for the first 7 days of treatment was 5.8 mg/day (n=165), which had increased to 8.5 mg/day (n=134) at week 3 and to 7.4 mg/day (n=96) up to end of week 12. On the day prior to the week 3 (LOCF) YMRS assessment, 53% of aripiprazole-treated patients received 30 mg/day, 42% received 15 mg/day, 5% received no dose and 1% received 45 mg/day. On the day prior to the week 12 (LOCF) YMRS assessment, the dose distribution was similar: 48% of the aripiprazole group received 30 mg/day, 42% received 15 mg/day, 9% received no dose and 1% received 45 mg/day. For haloperidol, the dose distribution at week 3 (LOCF) was 50%, 28%, 17% and 5% receiving 5 mg/day, 10 mg/day, 15 mg/day and no dose respectively. Similar dose distribution was seen at week 12 (LOCF) with 59%, 16%, 17% and 9% of patients receiving 5 mg/day, 10 mg/day, 15 mg/day and no dose respectively.
During the first 3-week treatment period, 72.9% of aripiprazole-treated patients, 77.0% of haloperidol-treated patients and 66.0% of placebo-treated patients received a concomitant medication active on the central nervous system. The most frequently used concomitant medications were anxiolytics (aripiprazole group 65.1%; haloperidol group 53.3%; placebo group 58.8%), followed by other analgesics and antipyretics (aripiprazole 26.5%; haloperidol 26.7%; placebo 27.5%). Similar rates were observed up to week 12. Use of anticholinergic medications was higher with haloperidol than aripiprazole up to the end of week 3 (aripiprazole group 15.1%; haloperidol group 44.2%; placebo group 6.5%) and up to the end of week 12 (aripiprazole 16.9%; haloperidol 50.3%), as was use of all concomitant medications used for the potential treatment of extrapyramidal symptoms up to the end of week 12 (aripiprazole 18.1%; haloperidol 52.7%).
Efficacy
Primary end-point
Aripiprazole produced statistically significantly greater improvements in the mean change from baseline to week 3 on the YMRS Total score when compared with placebo (–12.0 v. −9.7; P=0.039; LOCF) (Fig. 2 and online Table DS2). Similar clinical improvements were also seen for haloperidol when compared with placebo (–12.8 v. −9.7; P=0.005; LOCF). Both aripiprazole (P=0.047) and haloperidol (P=0.030) demonstrated statistically significantly greater improvements in YMRS Total score as early as day 2 (Fig. 2). Improvements beyond day 2 were statistically significantly greater for aripiprazole at weeks 2 and 3 and showed numerically greater improvement at days 4, 7 and 10. Haloperidol produced statistically significant improvements at all post-baseline assessments.
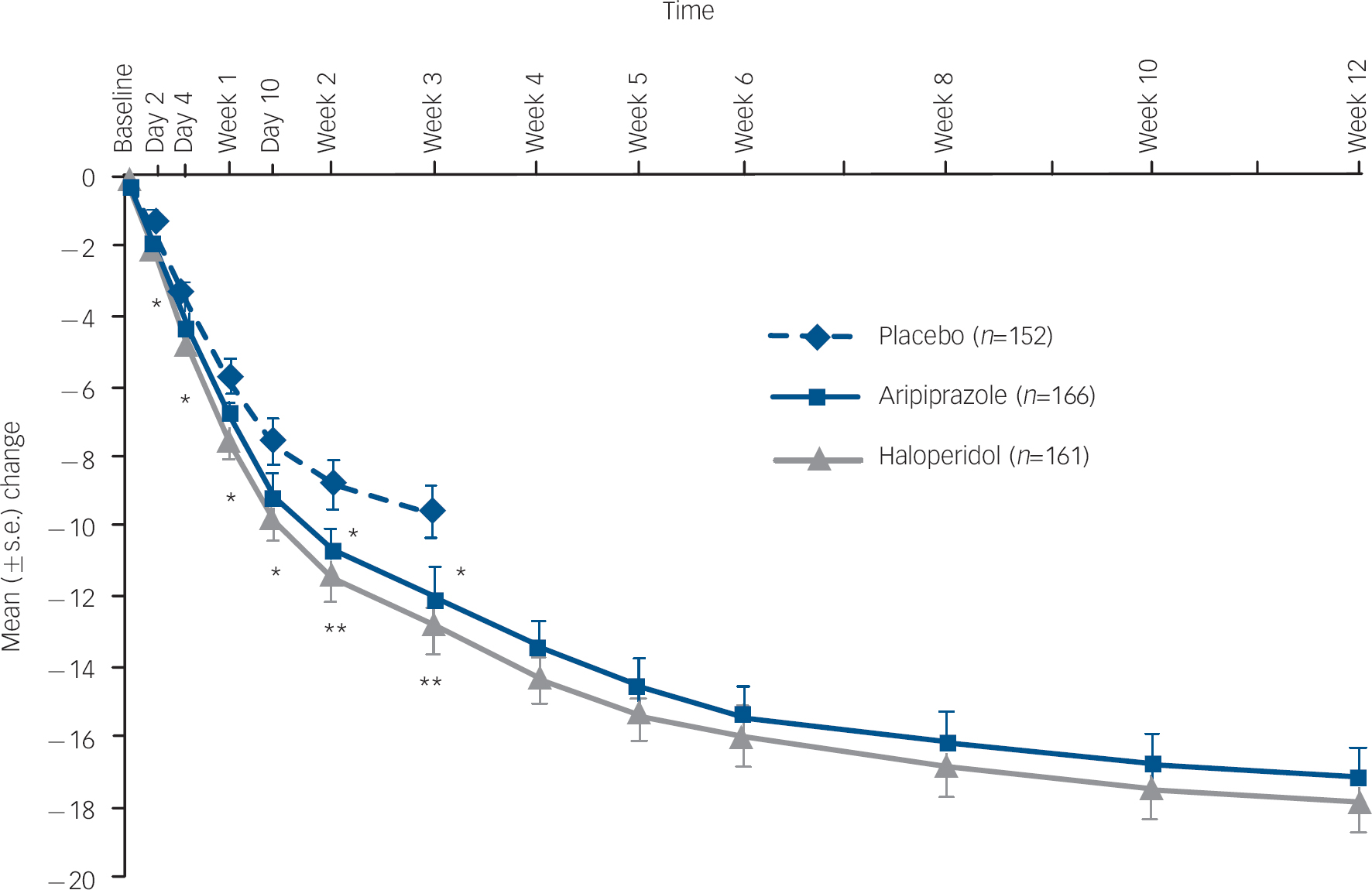
Fig. 2 Mean change (s.e.) from baseline in Young Mania Rating Scale (YMRS) Total score, efficacy sample (last observation carried forward).a a. Baseline YMRS scores: placebo, 28.8; haloperidol, 28.0; aripiprazole, 28.4. *P≤0.05; **P≤0.01 v. placebo.
For patients with bipolar I disorder manic type experiencing a manic episode (placebo n=124; aripiprazole n=132; haloperidol n=133), improvements in the mean change from baseline to week 3 on the YMRS Total score were greater for aripiprazole (–11.9) and haloperidol (–12.8) than with placebo (–8.7). The treatment difference was −3.3 (95% CI −5.7 to −0.8) for aripiprazole–placebo and −4.1 (95% CI −6.6 to −1.6) for haloperidol–placebo.
Secondary end-points
At week 3, the mean change from baseline in CGI–BP Severity of Illness (Mania) score was statistically significantly greater with aripiprazole than placebo (–1.4 v. −1.2, P=0.044; LOCF) (Fig. 3). There was also a statistically significant reduction in CGI–BP Severity of Illness (Mania) score in haloperidol-treated patients compared with placebo at week 3 (–1.6 v. −1.2, P=0.004; LOCF).
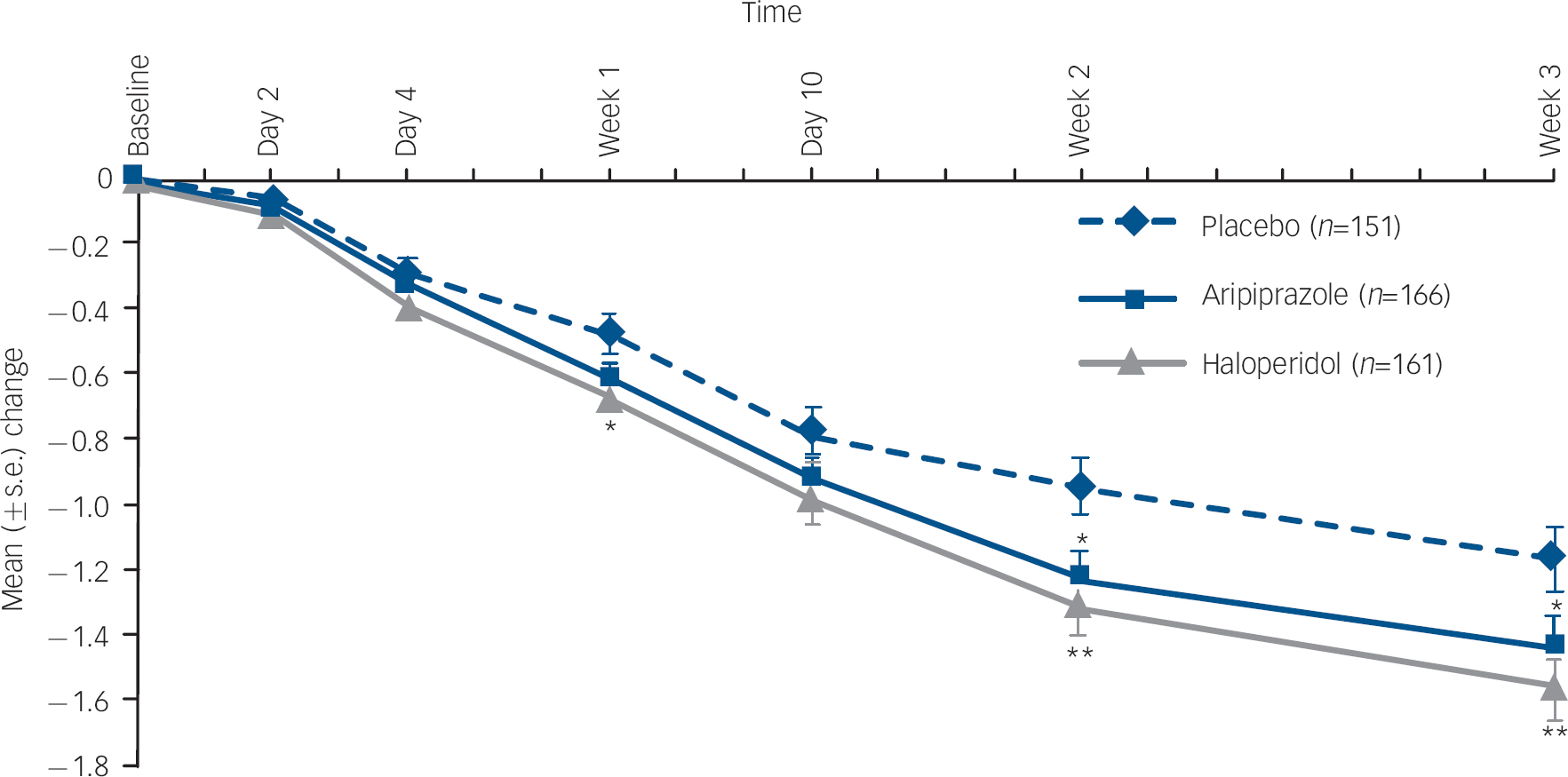
Fig. 3 Mean change (s.e.) from baseline Clinical Global Impression – Bipolar version (CGI–BP) Severity of Illness (Mania) score, efficacy sample (last observation carried forward).a. a. Baseline CGI–BP scores: placebo, 4.6; haloperidol, 4.5; aripiprazole, 4.5. *P≤0.05; **P≤0.01 v. placebo.
Improvements from baseline in YMRS Total score were maintained to week 12 (LOCF) for both aripiprazole (–17.2) and haloperidol (–17.8; P=0.564). Improvements from baseline in CGI–BP Severity of Illness (Mania) score were also maintained to week 12 (LOCF) for aripiprazole (–2.1) and haloperidol (–2.2) (Fig. 4).
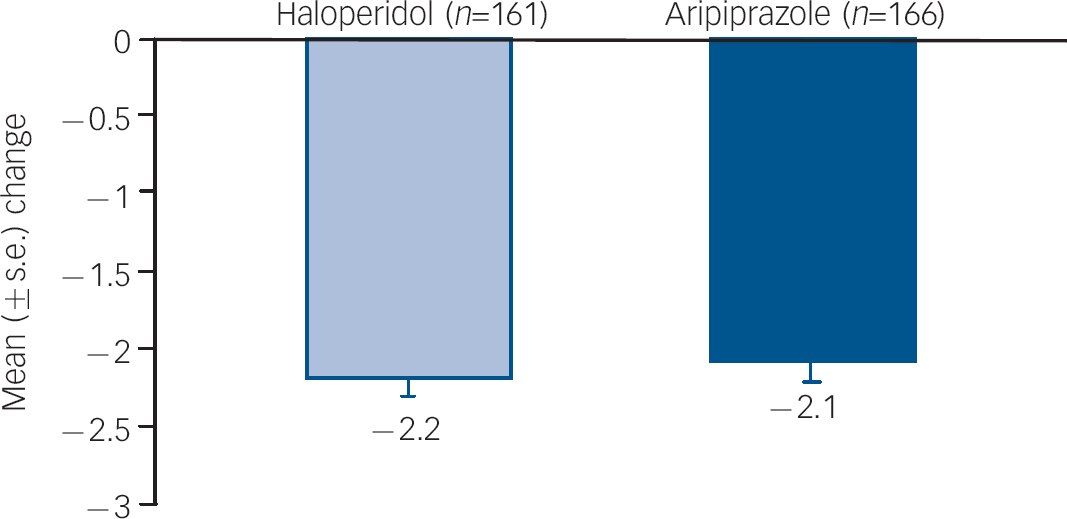
Fig. 4 Mean change from baseline to week 12 in CGI–BP Severity (Mania) score, efficacy sample (LOCF).a CGI–BP, Clinical Global Impression – Bipolar version; LOCF, last observation carried forward. a. Baseline CGI–BP scores: haloperidol, 4.5; aripiprazole, 4.5.
Response rates at week 3 (LOCF) were greater with aripiprazole (47.0%) and haloperidol (49.7%) than with placebo (38.2%), although neither was statistically significant (P=0.145 and P=0.069 respectively). Response rates at week 12 (LOCF) increased with both aripiprazole (72.3%) and haloperidol (73.9%) (P=0.909; ratio of response rates 1.0, 95% CI 0.9–1.1; number needed to treat (NNT) haloperidol v. aripiprazole=63). Remission rates at week 3 (LOCF) were also greater with aripiprazole (44.0%) and haloperidol (45.3%) v. placebo (36.8%), although neither was statistically significant (P=0.242 and P=0.206 respectively). By week 12, remission rates had increased to 69.9% with aripiprazole and 71.4% with haloperidol (P=0.896; ratio of remission rates 1.0, 95% CI 0.9–1.2; NNT haloperidol v. aripiprazole=67).
Table 1 summarises the findings of other secondary efficacy measures. Mean change from baseline in CGI–BP Severity of Illness (Overall) score was statistically significantly improved with aripiprazole v. placebo at week 3. Mean CGI–BP Change from Preceding Phase (Mania) score was also statistically significant in favour of aripiprazole at week 3. Aripiprazole produced statistically significant improvements for mean change from baseline in PANSS Total score, and Positive, Cognitive and Hostility sub-scale scores compared with placebo at week 3. All improvements seen with aripiprazole treatment were maintained, or improved, at week 12 and were similar to those seen with haloperidol. At week 3, changes from baseline in the CGI–BP Severity of Illness (Depression) score and MADRS scores showed similar small improvements in the haloperidol and aripiprazole treatment arms compared with placebo, and the improvements in these depression scales were maintained to end-point (week 12). There was no significant difference in the rate of emergent depression (MADRS Total score ≥18 plus ≥4-point increase from baseline for any two consecutive assessments) between either aripiprazole (6.0%) or haloperidol (1.9%) v. placebo (4.6%) at week 3 (P=0.575 and P=0.169 respectively). At week 12, rates of emergent depression were 4.3% for haloperidol and 9.6% for aripiprazole (ratio of incidence rates 2.2, 95% CI 0.9–5.3).
Table 1 Secondary efficacy end-points from baseline to weeks 3 and 12 (last observation carried forward)

| Rating scale | Placebo (n=151), mean (s.e.) | Haloperidol (n=161), mean (s.e.) | Aripiprazole (n=166), mean (s.e.) |
|---|---|---|---|
| CGI—BP Severity of Illness score | |||
| Overall | |||
| Baseline | 4.5 (0.1) | 4.4 (0.1) | 4.5 (0.1) |
| Change from baseline to week 3 | -1.1 (0.1) | -1.5 (0.1)** | -1.4 (0.1)* |
| Change from baseline to week 12 | — | -2.0 (0.1) | -1.9 (0.1) |
| Depression | |||
| Baseline | 1.5 (0.1) | 1.5 (0.1) | 1.7 (0.1) |
| Change from baseline to week 3 | -0.2 (0.1) | -0.1 (0.1) | -0.1 (0.1) |
| Change from baseline to week 12 | — | -0.1 (0.1) | -0.0 (0.1) |
| Change from Preceding Phase (Mania) | |||
| Change from baseline to week 3 | 3.0 (0.1) | 2.6 (0.1)** | 2.7 (0.1)* |
| Change from baseline to week 12 | — | 2.3 (0.1) | 2.3 (0.1) |
| MADRS Total Score a | |||
| Baseline | 8.1 (0.3) | 8.1 (0.3) | 8.3 (0.3) |
| Change from baseline to week 3 | -2.1 (0.4) | -2.5 (0.4) | -1.8 (0.4) |
| Change from baseline to week 12 | — | -2.4 (0.5) | -1.7 (0.5) |
| PANSS score b | |||
| Total | |||
| Baseline | 54.4 (0.8) | 54.1 (0.8) | 54.8 (0.8) |
| Change from baseline to week 3 | -4.7 (1.0) | -8.8 (1.0)** | -8.2 (1.0)* |
| Change from baseline to week 12 | — | -11.7 (1.1) | -9.8 (1.1) |
| Positive sub-scaleb | |||
| Baseline | 16.4 (0.4) | 16.1 (0.3) | 16.0 (0.3) |
| Change from baseline to week 3 | -2.4 (0.4) | -4.2 (0.4)*** | -3.8 (0.4)* |
| Change from baseline to week 12 | — | -5.4 (0.4) | -4.9 (0.4) |
| Negative sub-scaleb | |||
| Baseline | 9.4 (0.2) | 9.5 (0.2) | 9.6 (0.2) |
| Change from baseline to week 3 | -0.1 (0.2) | -0.3 (0.2) | -0.4 (0.2) |
| Change from baseline to week 12 | — | -0.3 (0.2) | -0.2 (0.2) |
| Cognitive sub-scaleb | |||
| Baseline | 14.9 (0.3) | 14.6 (0.3) | 14.7 (0.3) |
| Change from baseline to week 3 | -1.5 (0.3) | -2.5 (0.3)* | -2.4 (0.3)* |
| Change from baseline to week 12 | — | -3.9 (0.4) | -3.2 (0.4) |
| Hostility sub-scaleb | |||
| Baseline | 9.7 (0.2) | 9.4 (0.2) | 9.7 (0.2) |
| Change from baseline to week 3 | -1.2 (0.3) | -2.6 (0.3)*** | -2.3 (0.3)** |
| Change from baseline to week 12 | — | -3.5 (0.3) | -3.0 (0.3) |
For patients who were responders at week 3, the mean change from baseline in YMRS Total score and CGI–BP Severity of Illness (Mania) score showed that the effects of aripiprazole and haloperidol observed at week 3 were maintained through to end-point at week 12 (Table 2).
Table 2 Efficacy results up to end of week 12 (LOCF) for responders at week 3a

| Rating scale | Haloperidol (n=67), mean (s.e.) | Aripiprazole (n=67), mean (s.e.) |
|---|---|---|
| YMRS Total score (overall) | ||
| Baseline | 27.2 (0.7) | 28.3 (0.7) |
| Change from baseline to week 3 | -19.7 (0.5) | -20.6 (0.5) |
| Change from baseline to week 12 | -24.2 (0.5) | -24.8 (0.5) |
| CGI—BP Severity of Illness (Mania) score | ||
| Baseline | 4.5 (0.1) | 4.5 (0.1) |
| Change from baseline to week 3 | -2.4 (0.1) | -2.5 (0.1) |
| Change from baseline to week 12 | -3.2 (0.1) | -3.2 (0.1) |
There was a similar improvement across groups in the mean change in LIFE–RIFT Total score from baseline to week 3 (aripiprazole −2.1; haloperidol −1.8; placebo −1.4) or to week 12 (aripiprazole −3.1; haloperidol −2.8).
Post hoc MMRM analysis of change in YMRS Total scores from baseline produced similar results to LOCF analysis. Aripiprazole produced statistically significantly greater improvements in mean change from baseline to week 3 in YMRS Total score when compared with placebo (–13.3 v. −10.9, P=0.030; MMRM, observed case data-set), as did haloperidol compared with placebo (–14.2 v. −10.9, P=0.003; MMRM, observed case data-set).
Improvements from baseline in YMRS Total score were maintained to week 12 for both aripiprazole and haloperidol (–22.3 v. −22.7; MMRM, observed case data-set).
Safety
The most frequently reported treatment-emergent adverse events during the study are shown in online Table DS3. The adverse event profiles of both treatments were similar during the acute treatment phase and the whole study, which included the maintenance of effect treatment phase. During acute treatment (weeks 1–3), the three most commonly occurring adverse events with aripiprazole were insomnia (13.9%), akathisia (9.0%) and extrapyramidal disorder (7.2%), and these occurred at a similar incidence during the 12 weeks of treatment (weeks 1–12): insomnia (14.5%), akathisia (11.4%), extrapyramidal disorder (7.8%). The three most common adverse events with haloperidol during acute treatment (weeks 1–3) and during the whole study (weeks 1–12) respectively, were akathisia (20.6% v. 24.8%), extrapyramidal disorder (12.1% v. 15.2%) and muscle rigidity (7.9% v. 9.7%). The incidence of adverse events related to extrapyramidal symptoms was 53.3% in the haloperidol group compared with 23.5% in the aripiprazole group (weeks 1–12). Akathisia was cited as a reason for study discontinuation in 1 aripiprazole-treated patient (0.6%) and 2 haloperidol-treated patients (1.2%), whereas extrapyramidal disorder was cited as a reason for discontinuation in 1 aripiprazole-treated patient (0.6%) and 4 haloperidol-treated patients (2.4%); these aripiprazole discontinuations occurred before week 3. During the 12 weeks of treatment, akathisia, at its maximum intensity, was generally reported as mild (aripiprazole n=7/19; haloperidol n=18/41) or moderate (aripiprazole n=10/19; haloperidol n=21/41) in severity; few patients reported akathisia as severe/very severe (aripiprazole n=2/19; haloperidol n=2/41). For patients in the aripiprazole group who reported akathisia as an adverse event, with a maximum severity of mild, moderate, severe or very severe, the corresponding median highest BARS global clinical assessment scores were 2, 2, 1 and 4 respectively. For patients in the haloperidol group who reported akathisia with a maximum severity of mild, moderate or severe, the corresponding median highest BARS global clinical assessment scores were 2, 2 and 3.5 respectively. Furthermore, in the majority of cases akathisia had its onset in the first 3 weeks of treatment (aripiprazole n=14/19; haloperidol n=31/41). For patients that reported akathisia, it had resolved by the last study visit (week 12) in 12/19 (63.2%) of aripiprazole-treated patients and 22/41 (53.7%) of haloperidol-treated patients, although data on resolution were missing for 1 (5.3%) and 9 (22.0%) of aripiprazole and haloperidol-treated patients respectively. For those participants whose akathisia resolved during the study, 1 (8.3%) aripiprazole-treated patient and 1 (4.5%) haloperidol-treated patient received a dose reduction and use of concomitant medication, 1 (4.5%) haloperidol-treated patient discontinued therapy and received concomitant medication, 9 (75.0%) aripiprazole-treated patients and 17 (77.3%) haloperidol-treated patients received concomitant medication only, and no intervention was provided in the remaining 2 (16.7%) aripiprazole-treated and 3 (13.6%) haloperidol-treated patients.
Serious adverse events were reported in 19 (11.4%) aripiprazole-treated patients and 5 (3%) haloperidol-treated patients. Fourteen (8.4%) of the aripiprazole-treated patients experienced at least one serious adverse event categorised as ‘psychiatric disorder’; one patient reported two psychiatric serious adverse events. Overall, there were four reports of mania (two worsening of bipolar mania and two relapse of manic symptoms), three reports of bipolar I disorder (one depressed episode, one worsening of manic episode and one worsening bipolar mania), three of bipolar disorder (one exacerbation of symptoms of bipolar disorder, one worsening of bipolar affective disorder (manic phase) and one worsening of bipolar disorder), two of depression, and one each of insomnia, psychotic disorder and suicide attempt (non-fatal).
At week 3, there was a statistically significantly greater mean change from baseline in the SAS Total score in the haloperidol group compared with the placebo group (1.7 v. −0.2, P<0.001; LOCF). The mean change in the aripiprazole group was not statistically significantly different from placebo (0.2 v. −0.2, P=0.163;LOCF). At week 12, aripiprazole had a statistically significant smaller mean change from baseline in SAS Total score than haloperidol (0.2 v. 1.3, P<0.001; LOCF). Similar changes were observed for BARS global assessment scores, with a significantly greater mean change in the haloperidol group at week 3 compared with placebo (0.4 v. −0.1, P<0.001; LOCF), whereas the change in the aripiprazole group was not significantly different from placebo (0.1 v. −0.1, P=0.060; LOCF). At week 12, there was also a statistically significant difference in the mean change from baseline in BARS score between aripiprazole and haloperidol (0.3 v.0.1, P<0.001; LOCF). At week 3, there were minimal changes from baseline in the mean AIMS Total score for the aripiprazole and placebo groups (0.1 v. 0.0, P=0.522; LOCF), whereas haloperidol was associated with statistically significant increases compared with placebo (0.5 v. 0.0, P=0.004; LOCF). However, there was no statistically significant treatment difference in the AIMS Total score between aripiprazole and haloperidol at week 12 (0.2 v.0.6, P=0.107; LOCF).
Weight gain
The mean change in weight from baseline to week 3 (LOCF) was not significantly different between placebo (0.4 kg, s.e.=0.3) and aripiprazole (0.2 kg, s.e.=0.3; P=0.703) or haloperidol (0.5 kg, s.d.=0.3; P=0.684). Mean change in weight from baseline to week 12 (LOCF) was also not significantly different between aripiprazole and haloperidol (0.70 kg, s.e.=0.3 v. 0.56 kg, s.e.=0.3; P=0.688, LOCF). The mean change in weight from baseline to week 3 (observed case analysis) was similar between placebo (0.5 kg, s.e.=0.3, n=115), aripiprazole (0.3 kg, s.e.=0.3, n=139) and haloperidol (0.5 kg, s.e.=0.3, n=127), as was weight change at week 12 (observed case analysis) between aripiprazole (1.0 kg, s.e.=0.3, n=96) and haloperidol (0.2 kg, s.e.=0.3, n=93). At week 3 (LOCF), the proportion of patients with clinically significant weight gain (≥7% from baseline) was also similar between placebo (9/140, 6.4%) and aripiprazole (3/156, 1.9%; P=0.065 v. placebo) and haloperidol (4/151, 2.6%; P=0.221 v. placebo). Incidence of clinically significant weight gain was also similar between aripiprazole and haloperidol at week 12 (5.1% v. 5.8%, P=0.723; LOCF). Observed case clinically significant weight gain produced similar findings.
Metabolics
Mean changes from baseline in metabolic parameters are shown in online Table DS4. There were no clinically meaningful differences between the treatment groups in the change from baseline in fasting glucose, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglycerides or total cholesterol levels at weeks 3 and 12.
No clinically meaningful differences in vital sign measurements were seen between treatment groups; no patients discontinued because of vital sign abnormalities. Fewer patients had potentially clinically relevant prolactin levels (>upper limit of normal) with aripiprazole (22.4%) v. haloperidol (66.2%) at week 12. At week 12, fewer aripiprazole- (12.8%) than haloperidol-treated patients (60.8%) had new-onset hyperprolactinaemia (baseline prolactin levels ≤upper limit of normal and week 12 prolactin levels >upper limit of normal). Aripiprazole-treated patients experienced a decrease in mean serum prolactin levels from baseline over 12 weeks, whereas mean serum prolactin levels increased with haloperidol (–13.4 ng/ml v. +6.7 ng/ml; P<0.001).
Discussion
Results from this large, randomised placebo-controlled double-blind study show that aripiprazole and haloperidol were both more effective than placebo for the acute and maintenance of effect treatment of bipolar disorder. Both aripiprazole and haloperidol resulted in early improvements in symptoms of acute mania and produced significant improvements over placebo on the primary efficacy end-point, mean change in YMRS Total score from baseline at week 3. Consistent with the significant improvements seen in the primary efficacy measure, aripiprazole also produced significant improvements over placebo on the key secondary efficacy measure, CGI–BP Severity of Illness (Mania) score at week 3, and several other secondary efficacy outcomes (CGI–BP Severity of Illness (Overall) and Change from Preceding Phase (Mania) scores, PANSS Total score and PANSS Positive, Cognitive and Hostility sub-scale scores). At week 3, response and remission rates for either active treatment arm were greater than for placebo but not significantly different. Improvements seen with aripiprazole and haloperidol at week 3 were maintained up to week 12. Although this study was not powered to compare the efficacy of aripiprazole and haloperidol, the magnitudes of improvement seen with each agent appear similar. Pharmacologically, the results of this study are interesting as they demonstrate that aripiprazole, a partial agonist at D2 receptors, confers similar antimanic effects to haloperidol, a D2 receptor antagonist.
Results from this study confirm findings from previous studies that have shown that aripiprazole is effective at rapidly controlling the symptoms of acute mania, Reference Keck, Marcus, Tourkodimitris, Ali, Liebeskind and Saha6,Reference Sachs, Sanchez, Marcus, Stock, McQuade and Carson7 including agitation, Reference Sachs, Gaulin, Gutierrez-Esteinou, McQuade, Pikalov and Pultz20 and adds to evidence supporting the use of aripiprazole monotherapy as maintenance of effect treatment beyond the acute phase. Reference Vieta, Bourin, Sanchez, Marcus, Stock and McQuade21,Reference Keck, Orsulak, Cutler, Sanchez, Torbeyns and Marcus22 In previous studies, aripiprazole was started at 30 mg/day, Reference Keck, Marcus, Tourkodimitris, Ali, Liebeskind and Saha6,Reference Sachs, Sanchez, Marcus, Stock, McQuade and Carson7 whereas this study demonstrated the efficacy of aripiprazole with a treatment regimen starting at 15 mg/day. As 42% of aripiprazole-treated patients were on 15 mg/day at week 3, this study demonstrated that a treatment regimen with a starting dose of 15 mg/day can be an effective approach for treating patients with acute bipolar mania.
The findings reported here are similar to results from 12-week studies with other atypical agents – olanzapine, Reference Tohen, Goldberg, Gonzalez-Pinto Arrillaga, Azorin, Vieta and Hardy-Bayle23 quetiapine Reference Bowden, Grunze, Mullen, Brecher, Paulsson and Jones24,Reference McIntyre, Brecher, Paulsson, Huizar and Mullen25 and risperidone Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26 – that have shown maintenance of effect for up to 12 weeks.
Although both response and remission rates at week 3 were numerically higher with both aripiprazole and haloperidol than placebo, statistical significance between either active treatment and placebo was not reached. It is likely that this is, in part, due to the high placebo response (38%) and remission (37%) rates, which are not uncommon in trials involving patients with bipolar disorder. Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26–Reference Tohen, Jacobs, Grundy, McElroy, Banov and Janicak28 Importantly, response and remission rates were comparable between aripiprazole and haloperidol at week 3 and showed continued improvement to week 12; at week 12, nearly 75% of patients had responded to treatment and the majority of patients had reached remission. Response rates reported here with aripiprazole at week 3 are similar to those seen in similar trials with risperidone (risperidone 48% v. placebo 33%) Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26 and quetiapine (quetiapine 43% v. placebo 35%), although response rates for haloperidol in this second study were higher (56%). Reference McIntyre, Brecher, Paulsson, Huizar and Mullen25 Furthermore, although completion rates reported here were similar between treatment arms at week 3, similar completion rates between treatment arms have also been observed in previous studies of this design with risperidone (risperidone 89%; placebo 85%; haloperidol 90%). Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26
There was a low rate of emergent depression over the 12-week study for both aripiprazole- and haloperidol-treated patients. Furthermore, MADRS scores, although low at baseline, showed no worsening of depressive symptoms with either treatment at week 3 and week 12, suggesting that improvement in mania was not associated with a worsening of depression in patients with mania.
The mean dosing of haloperidol in this study (week 3: 8.5 mg/day; week 12: 7.4 mg/day) was comparable with haloperidol dosing in similar 12-week studies using other atypical antipsychotic agents; in one study, mean haloperidol dosing ranges were from 9.6 mg/day (week 1) to 5.2 mg/day (week 12), Reference Tohen, Goldberg, Gonzalez-Pinto Arrillaga, Azorin, Vieta and Hardy-Bayle23 whereas in a second study mean haloperidol dosing ranges were from 7.4 mg/day (week 12) to 8 mg/day (week 3). Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26
Tolerability
Aripiprazole was generally well tolerated over the entire 12-week study, with an adverse event profile similar to that seen in previous short-term Reference Keck, Marcus, Tourkodimitris, Ali, Liebeskind and Saha6,Reference Sachs, Sanchez, Marcus, Stock, McQuade and Carson7 and long-term studies. Reference Keck, Calabrese, McQuade, Carson, Carlson and Rollin8,Reference Keck, Calabrese, McIntyre, McQuade, Carson and Eudicone9 The incidence of adverse events related to extrapyramidal symptoms with haloperidol was more than double that with aripiprazole, as was the use of concomitant medications for the potential treatment of such symptoms. Objective measurement of extrapyramidal symptoms using the SAS and BARS global assessment rating scales also showed a significantly lower risk of such symptoms with aripiprazole than with haloperidol. Higher rates of adverse events related to extrapyramidal symptoms with haloperidol are a well-known side-effect of treatment with typical antipsychotics and represent an important factor when considering long-term treatment of patients with such bipolar mania, as patients tend to be more at risk for antipsychotic-induced tardive dyskinesia than patients with schizophrenia. Reference Nasrallah, Churchill and Hamdan-Allan29,Reference Kane and Smith30 For those patients who experienced akathisia, this was manageable, generally mild to moderate in severity and resolved before the end of the study in both treatment groups. It is curious to consider whether all treatment-emergent akathisia events are actually true akathisia. Akathisia, as currently defined in clinical trials nomenclature, might be capturing clinical features related to other syndromes such as activation or anxiety symptoms.
Aripiprazole also demonstrated a low propensity to elevate prolactin levels compared with haloperidol. Fewer patients receiving aripiprazole experienced prolactin levels above the upper limit of normal compared with those receiving haloperidol; mean serum prolactin levels decreased with aripiprazole, whereas they increased with haloperidol.
Minimal mean changes in body weight were seen with aripiprazole and haloperidol over the 12-week study period, and the incidence of clinically significant weight gain with aripiprazole was comparable with placebo at week 3, and remained low at week 12. This is an important finding given that patients with bipolar disorder are at increased risk of obesity and associated medical morbidity than the general population. Reference McIntyre, Konarski, Soczynska, Wilkins, Panjwani and Bouffard31 Indeed, the average BMI of patients in this study was high (∼27 kg/m2), and approximately 40% of patients had a BMI >27 kg/m2, emphasising the importance of lack of weight gain when choosing an atypical antipsychotic for long-term treatment. Aripiprazole also showed a low potential for metabolic effects, with minimal effects on lipid and glucose levels as seen in longer-term trials in bipolar mania. Reference Keck, Calabrese, McQuade, Carson, Carlson and Rollin8,Reference Keck, Calabrese, McIntyre, McQuade, Carson and Eudicone9
Methodological considerations
Findings of this study are strengthened by the large patient population and the randomised placebo-controlled design. Although the lack of a placebo comparator arm beyond the first 3 weeks of study could be considered a limitation, use of placebo for only 3 weeks is an accepted methodology and a previous study with risperidone used a similar design. Reference Smulevich, Khanna, Eerdekens, Karcher, Kramer and Grossman26 Furthermore, there are many ethical arguments against the use of placebo beyond the acute treatment phase in patients with bipolar disorder. Reference Vieta and Carne27
In conclusion, aripiprazole significantly improved symptoms during the 3-week placebo-controlled phase of the study in patients with acute mania. The beneficial effects of aripiprazole were sustained through to week 12 and were similar to haloperidol, a gold-standard antipsychotic treatment. Aripiprazole was generally well tolerated, with a lower incidence of extrapyramidal symptoms v. haloperidol.
Acknowledgements
This study was supported by Bristol-Myers Squibb (Princeton, New Jersey) and Otsuka Pharmaceutical Co., Ltd (Tokyo, Japan). Editorial support for the preparation of this manuscript was provided by Michelle O'Donovan, PhD, Ogilvy Healthworld Medical Education. Funding was provided by Bristol-Myers Squibb.
The authors would like to acknowledge the principal investigators of this study, CN138-162: Kausar Yakhin, MD, Kazan State Medical University, Russian Federation; Yury Aleksandrovskiy, MD, Serbsky National Research Center for Social and Forensic Psychiatry, Russian Federation; Margarita Morozova, MD, Municipal Clinical Psychiatry Hospital #14, Russian Federation; Anatoly Smulevich, MD, Hospital #1 Moscow, Russian Federation; Galina Panteleyeva, MD, Mental Health Research Center, Moscow, Russian Federation; Boris Andreyev, MD, Kashenko 1st Stp City Mental Hospital, Russian Federation; Yury Suchkov, MD, Municipal Hospital #1, Russian Federation; Sergey Elkin, MD, Psychiatric Hospital #3, Russian Federation; Pavo Filaković, MD, Clinical Hospital Osijek, Croatia (Hrvatska); Vera Folnegović-Šmalc, MD, PhD, Psychiatric Hospital Vrapće, Croatia (Hrvatska); Dulijano Ljubicic, MD, PhD, Ljiljana Moro, MD (Prev), Clinical Hospital Centre Rijeka, Croatia (Hrvatska); Goran Dodig, MD, Clinical Hospital Split, Croatia (Hrvatska); Michael Burducovsky, MD, Psychiatric Hospital #4, Russian Federation; José de Jesús Castillo, MD, Centros Avanzados En Salud Animica, Mexico; Juan-Ignacio Rosales, MD, Clinica San Rafael, Mexico; Wazcar Verduzco, MD, Hospital de Psiquiatría de San, Mexico; Rogelio Apiquián, MD, Grupo Médico Carracci, Mexico; Raymond Manning, MD, CNRI – Los Angeles, California, USA; Jorge Porras, MD, Gregory S. Paniccia, MD (Prev), David M. Marks, MD (Prev), eStudySite, San Diego, USA; Adam Lowy, MD, Comprehensive NeuroScience Inc, Washington, USA; Mark Scheier, MD, Bum Soo Lee, MD (Prev), Anaheim Research Center, LLC, California, USA; Rajinder Shiwach, MD, InSite Clinical Research, DeSoto, USA; Mohammed Bari, MD, Synergy Clinical Research Center, San Diego, USA; Richard Knapp, MD, CORE Research Inc, Maitland, USA; Morteza Marandi, MD, David Sack, MD (Prev), Comprehensive NeuroScience, Cerritos, USA; Michael Plopper, MD, Sharp Mesa Vista Hospital, San Diego, USA; Pauline Powers, MD, University of South Florida, USA; Chandra S. Krishnasastry, MD, Clinical Research Services at Tennessee Christian Medical Center, USA; Kashinath G. Yadalam, MD, Lake Charles Clinical Trials, USA; G. Michael Dempsey, MD, Albuquerque Neuroscience Inc, USA; Prashant Gajwani, MD, Mood Disorders Program, Cleveland, USA; Gunnar L. Larson, MD, CCRI; Clement J. Zabocki, VA Medical Center, Milwaukee, USA; Paresh Ramjee, MBCHB, Vista Private Clinic, Pretoria, South Africa; Farouk Randeree, MBCHB, 302 Musgrave Park, Durgan, South Africa; Cornelius Van Staden, MBCHB, Weskoppies Hospital, Pretoria West, South Africa; Roberto Gastiaburu, MD, Hospital De Policia, Lima, Peru; Abel Sagastegui, MD, Asociacion De Lucha Contra El Estigma, Lima, Peru; Pablo Adan, MD, Hospital Nacional Edgardo Rebagliati Martins, Lima, Peru; Boris Tsygankov, MD, Moscow State University of Medicine and Dentistry, Moscow, Russian Federation; Natalia Dobrovolskaya, MD, City Psychoneurological Dispensary #7 with Hospital, St Petersburg, Russian Federation; Luchezar Hranov, MD, II Psychiatric Clinic St. Naum Psych Hosp, Sofia, Bulgaria; Loris Sayan, MD, PhD, Psychiatry Dispensary DDPDS Prof., Bulgaria; Stefan Todorov, MD, PhD, First Psychiatric Clinic-MHAT ‘St Marina’, Bulgaria; Vihra Milanova, MD, Alexander University Hospital, Sofia, Bulgaria; Vihra Milanova, MD, Alexander University Hospital, Sofia, Bulgaria; Salumu Selemani, MBCHB, Biothuso Care Givers, South Africa; Mikhail Sheyfer, MD, Samara Psychiatric Hospital, Russian Federation; Pepa Dimitrova, MD, MHAT Psychiatric Clinic, Pleven, Bulgaria; Ivan Gerdjikov, MD, I-St Maleand I-St Female Psychiatry Clinics, Bulgaria; Svetlozar Haralanov, MD, II Psychiatric Clinic St. Naum Psych Hosp, Sofia, Bulgaria; Amado Nieto, MD, Clinica Psiquiátrica Everardo Neuman Peña, Mexico; Alfonso Ontiveros, MD, Hospital San José Tec de Monterrey, Monterrey, Mexico; Pavel Sidorov, MD, PhD, Northern State Medical University, Russian Federation; Andrey Gribanov, MD, Lipetsk Regional Psychiatric and Neurologic Hospital #1, Lipetsk, Russian Federation; Alexander Bukhanovskiy, MD, PhD, Scientific Center for Treatment and Rehabilitation ‘Phoenix’, Russian Federation; Sergio J Villaseñor, MD, Hospital Civil ‘Fray Antonio Alcalde’, Mexico; Felipe Triay, MD, 35B # 473 x50 y 52, Fracc. Santa Cecilla, Mexico; Victor Vid, MD, Bekhterev Psychoneurological Research Institute, St Petersburg, Russian Federation; Sergey Kolov, MD, Volgograd State Medical Academy, Russian Federation; Francisco Brandi Rigal, MD, Comunidad Terapéutica, Mexico; J. Gary Booker, MD, 851 Olive Street, Shreveport, USA; Louise M. Beckett, MD, IPS Research Company, Oklahoma City, USA; Joachim Raese, MD, Behavioral Health 2000, LLC, Riverside, California, USA; Gregory F. Bishop, MD, eStudySite, San Diego, USA; David P. Walling, PhD, Collaborative Neuroscience Network Inc, Torrance, California, USA.








eLetters
No eLetters have been published for this article.