



Introduction
The motivation behind writing this review was to summarize the current views on biology of Trypanosomatidae. While several recent reviews were focused on specific aspects of this field (Hajduk and Ochsenreiter, Reference Hajduk and Ochsenreiter2010; Jackson, Reference Jackson2015; Read et al., Reference Read, Lukeš and Hashimi2016; Gibson, Reference Gibson, Archibald, Simpson and Slamovits2017; Kaufer et al., Reference Kaufer, Ellis, Stark and Barratt2017), a rather broad aim of our paper includes advances in taxonomy, genetics, molecular and cellular biology, and biochemistry of these fascinating organisms.
It is hard to overemphasize the significance of trypanosomatids for both basic and applied research. This group was first outlined by William Saville-Kent, who united genera Herpetomonas and Trypanosoma into the new order Trypanosomata (Saville-Kent, Reference Saville-Kent1880). This order (now spelled as Trypanosomatida) encompasses a single family Trypanosomatidae (Hoare, Reference Hoare1966; Vickerman, Reference Vickerman, Vickerman and Preston1976), the members of which are obligatory parasites of invertebrates, vertebrates and plants (Nussbaum et al., Reference Nussbaum, Honek, Cadmus and Efferth2010; Lukeš et al., Reference Lukeš, Skalický, Týč, Votýpka and Yurchenko2014). Dixenous (with two hosts in their life cycle) parasites employ an invertebrate (arthropod or leech) vector to shuttle between the vertebrate (genera Endotrypanum, Leishmania, Paraleishmania, Trypanosoma) or plant (genus Phytomonas) hosts. Most monoxenous (with a single host) trypanosomatids parasitize insects. The importance of dixenous species is incontestable as they cause severe diseases in humans, domestic animals and economically important plants (Simpson et al., Reference Simpson, Stevens and Lukeš2006). In contrast, until recently monoxenous species have been viewed as less relevant relatives of the all-important pathogens such as Trypanosoma brucei, T. cruzi or Leishmania spp. Only in the last decade had they attracted the due attention as the group of high diversity, ability to adapt to dramatically different environmental conditions, ubiquity and impact on insect hosts’ communities (Maslov et al., Reference Maslov, Votýpka, Yurchenko and Lukeš2013; Hamilton et al., Reference Hamilton, Votýpka, Dostalova, Yurchenko, Bird, Lukeš, Lemaitre and Perlman2015; Votýpka et al., Reference Votýpka, d'Avila-Levy, Grellier, Maslov, Lukeš and Yurchenko2015; Ishemgulova et al., Reference Ishemgulova, Butenko, Kortišová, Boucinha, Grybchuk-Ieremenko, Morelli, Tesařová, Kraeva, Grybchuk, Pánek, Flegontov, Lukeš, Votýpka, Pavan, Opperdoes, Spodareva, d'Avila-Levy, Kostygov and Yurchenko2017; Lukeš et al., Reference Lukeš, Butenko, Hashimi, Maslov, Votýpka and Yurchenko2018). Besides, as the dixenous trypanosomatids evolved from their monoxenous kin (Fernandes et al., Reference Fernandes, Nelson and Beverley1993; Hughes and Piontkivska, Reference Hughes and Piontkivska2003; Jirků et al., Reference Jirků, Yurchenko, Lukeš and Maslov2012; Flegontov et al., Reference Flegontov, Votýpka, Skalický, Logacheva, Penin, Tanifuji, Onodera, Kondrashov, Volf, Archibald and Lukeš2013), the study of insect parasites is imperative for understanding the evolutionary pathways in the family.
Classification system and evolution of dixenous lifestyle
The traditional classification system of this group relied on a very limited set of diagnostic traits and, in essence, was based on rough cell morphology and particularities of the life cycle, such as the monoxenous vs dixenous mode, as well as host specificity (Hoare and Wallace, Reference Hoare and Wallace1966; Vickerman, Reference Vickerman, Vickerman and Preston1976). A modern system is phylogeny based, but even after more than two decades of molecular phylogenetic analyses some relationships among the trypanosomatid major clades remain unresolved (Votýpka et al., Reference Votýpka, d'Avila-Levy, Grellier, Maslov, Lukeš and Yurchenko2015). Nowadays, nucleotide sequences with thousands of informative characters are routinely used for inferring evolutionary relationships between these protists (Borghesan et al., Reference Borghesan, Ferreira, Takata, Campaner, Borda, Paiva, Milder, Teixeira and Camargo2013; d'Avila-Levy et al., Reference d'Avila-Levy, Boucinha, Kostygov, Santos, Morelli, Grybchuk-Ieremenko, Duval, Votýpka, Yurchenko, Grellier and Lukeš2015). The main constraint of such a molecular phylogenetic approach is that it remains based on a limited number of genetic loci – usually 18S rRNA and gGAPDH (glycosomal glyceraldehyde 3-phosphate dehydrogenase). These molecular markers work well for resolving relationships between genera and higher taxa, but are not best-suited to delineate sub-generic ranks (Yurchenko et al., Reference Yurchenko, Lukeš, Xu and Maslov2006b; Votýpka et al., Reference Votýpka, Maslov, Yurchenko, Jirků, Kment, Lun and Lukeš2010). In any case, phylogenies inferred from single or a few genes can be misleading and frequently are poorly resolved (Grybchuk-Ieremenko et al., Reference Grybchuk-Ieremenko, Losev, Kostygov, Lukeš and Yurchenko2014; Yurchenko et al., Reference Yurchenko, Votýpka, Tesařová, Klepetková, Kraeva, Jirků and Lukeš2014; Frolov et al., Reference Frolov, Malysheva, Ganyukova, Yurchenko and Kostygov2017). The foreseeable solution to this problem (facilitated by rapidly decreasing prices of the next-generation sequencing and development of increasingly powerful methods of data analysis) lies with phylogenomic analyses based on the whole-genome sequences (Flegontov et al., Reference Flegontov, Butenko, Firsov, Kraeva, Eliáš, Field, Filatov, Flegontova, Gerasimov, Hlaváčová, Ishemgulova, Jackson, Kelly, Kostygov, Logacheva, Maslov, Opperdoes, O'Reilly, Sádlová, Ševčíková, Venkatesh, Vlček, Volf, Votýpka, Záhonová, Yurchenko and Lukeš2016; Skalický et al., Reference Skalický, Dobáková, Wheeler, Tesařová, Flegontov, Jirsová, Votýpka, Yurchenko, Ayala and Lukeš2017).
In the current classification system, six formally recognized subfamilies and 22 genera are included in the family Trypanosomatidae (Fig. 1). The subfamily Leishmaniinae (Jirků et al., Reference Jirků, Yurchenko, Lukeš and Maslov2012; Kostygov and Yurchenko, Reference Kostygov and Yurchenko2017) unites monoxenous parasites of insects (genera Borovskiya, Crithidia, Leptomonas, Lotmaria, Novymonas and Zelonia) and dixenous parasites of insects and vertebrates (genera Leishmania, Paraleishmania and ‘Endotrypanum’). The monogeneric subfamilies Blechomonadinae (Votýpka et al., Reference Votýpka, Suková, Kraeva, Ishemgulova, Duží, Lukeš and Yurchenko2013) and Paratrypanosomatinae (Flegontov et al., Reference Flegontov, Votýpka, Skalický, Logacheva, Penin, Tanifuji, Onodera, Kondrashov, Volf, Archibald and Lukeš2013) include monoxenous parasites of Siphonaptera (genus Blechomonas) and the early branching lineage of the genus Paratrypanosoma, respectively. The subfamily Strigomonadinae (Votýpka et al., Reference Votýpka, Kostygov, Kraeva, Grybchuk-Ieremenko, Tesařová, Grybchuk, Lukeš and Yurchenko2014) encompasses bacterial endosymbiont-harbouring genera Angomonas, Kentomonas, and Strigomonas, while the subfamily Phytomonadinae (Yurchenko et al., Reference Yurchenko, Kostygov, Havlová, Grybchuk-Ieremenko, Ševčíková, Lukeš, Ševčík and Votýpka2016) contains genera Phytomonas, Herpetomonas and Lafontella. Previously, the genus Trypanosoma was not assigned to any subfamily. We deem, that in accordance with the International Code of Zoological Nomenclature principle of coordination, this genus, as a name-bearing type, should be included into the nominotypical subfamily, i.e. Trypanosomatinae. The genera Blastocrithidia, Jaenimonas (Hamilton et al., Reference Hamilton, Votýpka, Dostalova, Yurchenko, Bird, Lukeš, Lemaitre and Perlman2015), Sergeia (Svobodová et al., Reference Svobodová, Zídková, Čepička, Oborník, Lukeš and Votýpka2007) and Wallacemonas (Kostygov et al., Reference Kostygov, Grybchuk-Ieremenko, Malysheva, Frolov and Yurchenko2014) remain orphans and not united into higher-order groups for now. In addition, several clades have been revealed by the analyses of environmental samples (Týč et al., Reference Týč, Votýpka, Klepetková, Šuláková, Jirků and Lukeš2013; Votýpka et al., Reference Votýpka, d'Avila-Levy, Grellier, Maslov, Lukeš and Yurchenko2015), yet their formal description awaits the availability of respective trypanosomatids in culture.
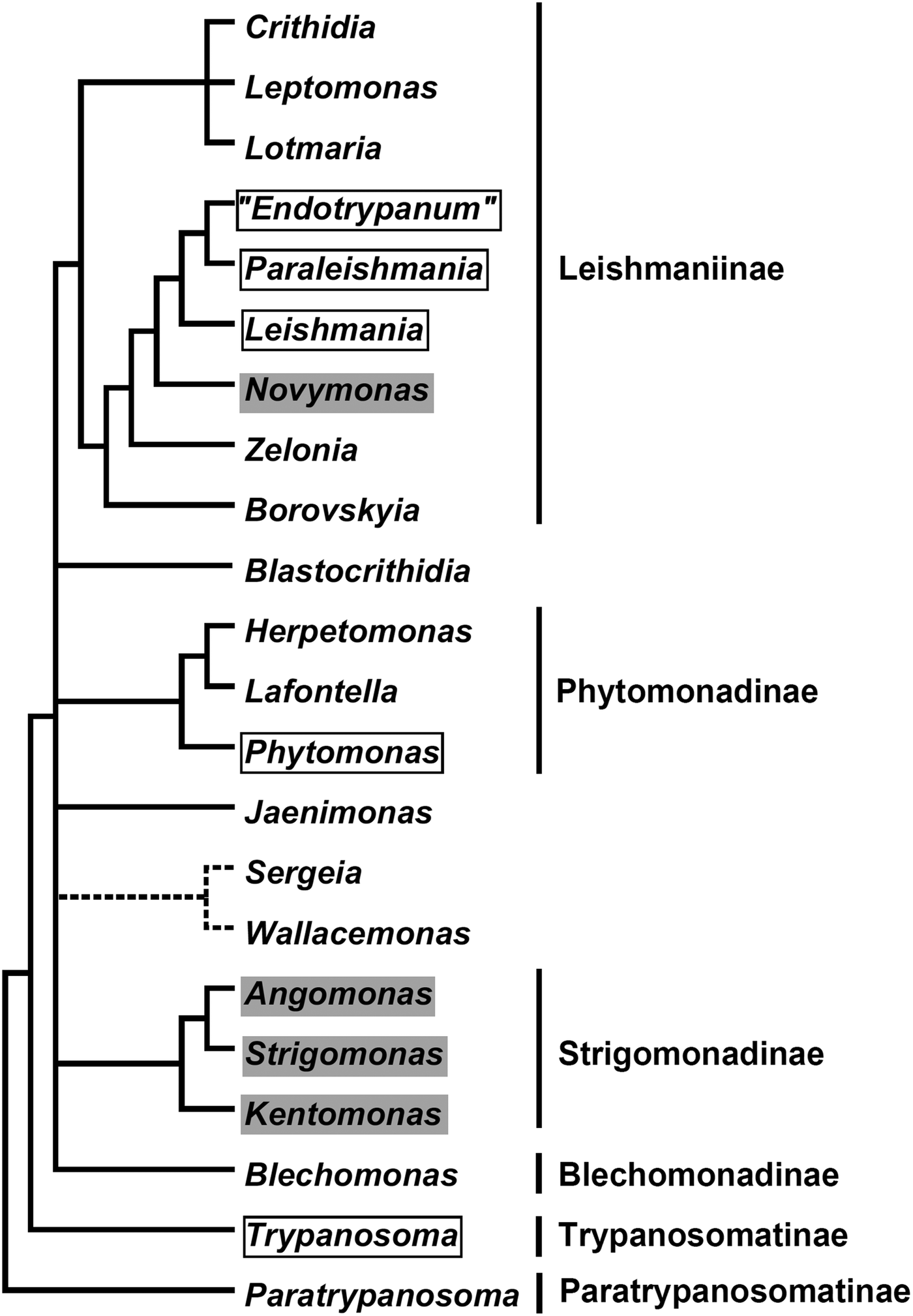
Fig. 1. A schematic phylogenetic tree demonstrating relationships between known Trypanosomatidae taxa. Dixenous and endosymbiont-containing genera are framed and shaded, respectively. The precise phylogenetic position of the Sergeia/Wallacemonas group is uncertain and indicated by broken lines. Formally recognized subfamilies are listed on the right.
The historical ‘chicken-or-egg’ debate over the origin of dixenous trypanosomatids (insect-first vs vertebrate-first scenarios) seems to be resolved. It is now generally accepted that the dixenous lifestyle has evolved from the monoxenous one several times in evolution leading to the emergence of the genera Trypanosoma, Leishmania, and Phytomonas (Fernandes et al., Reference Fernandes, Nelson and Beverley1993; Hamilton et al., Reference Hamilton, Stevens, Gaunt, Gidley and Gibson2004; Lukeš et al., Reference Lukeš, Skalický, Týč, Votýpka and Yurchenko2014). The boundary between monoxenous and dixenous types of parasitism appears to be dynamic, as members of some typical monoxenous groups were found in warm-blooded (usually, immuno-compromised) hosts (Dedet and Pratlong, Reference Dedet and Pratlong2000; Chicharro and Alvar, Reference Chicharro and Alvar2003; Ghosh et al., Reference Ghosh, Banerjee, Sarkar, Datta and Chatterjee2012), while descendants of some formerly dixenous species switched back to monoxeny (Lai et al., Reference Lai, Hashimi, Lun, Ayala and Lukeš2008; Frolov et al., Reference Frolov, Malysheva and Kostygov2016). The molecular mechanisms governing successful transitions between the monoxenous and dixenous life cycles remain to be investigated.
Nuclear gene organization and expression
The Excavata, a protist superclade, which includes trypanosomatids, had separated very early from the rest of the eukaryotic tree (Cavalier-Smith et al., Reference Cavalier-Smith, Chao, Snell, Berney, Fiore-Donno and Lewis2014). This long independent evolution resulted in the development of a wide range of cellular and molecular features unique to this group. Thus, while staying within the general eukaryotic cell and molecular layout, trypanosomatids had evolved to become drastically different from the ‘textbook’ eukaryotes, such as Metazoa or Fungi, at every level of gene organization and expression (Lukeš et al., Reference Lukeš, Skalický, Týč, Votýpka and Yurchenko2014). Naturally, these differences, especially those present in pathogenic trypanosomatids, have been a subject of intense investigations ultimately aimed at finding potential targets for disease treatment and control, as well as expanding the knowledge base beyond boundaries of the common model organisms. Since the sheer volume of the accumulated information precludes its in-depth review within the limits of a single section or even an entire review, we kindly refer the reader to the numerous reviews dealing with specific aspects of this burgeoning field (Myler, Reference Myler, Myler and Fasel2008; Bindereif, Reference Bindereif2012; Preusser et al., Reference Preusser, Jae and Bindereif2012; Clayton, Reference Clayton2014; Horn, Reference Horn2014). Below is a brief overview of the most striking features of trypanosomatid gene expression, which set these parasites very far apart from their metazoan hosts and vectors.
Genome organization
Trypanosomatid genomes are relatively compact, ranging from 18.0 Mb in Phytomonas sp. to 32.6 Mb in Crithidia fasciculata. The number of nuclear-encoded genes varies from the reduced set of 6400 genes in Phytomonas to 16 900 genes estimated for Angomonas (Porcel et al., Reference Porcel, Denoeud, Opperdoes, Noel, Madoui, Hammarton, Field, Da Silva, Couloux, Poulain, Katinka, Jabbari, Aury, Campbell, Cintron, Dickens, Docampo, Sturm, Koumandou, Fabre, Flegontov, Lukeš, Michaeli, Mottram, Szoor, Zilberstein, Bringaud, Wincker and Dollet2014; Jackson, Reference Jackson2015). The chromosomal structure is by far best known for T. brucei. Its genome is divided among 11 large diploid chromosomes (1 to 6 Mb in size) (Melville et al., Reference Melville, Leech, Gerrard, Tait and Blackwell1998), ~5 intermediate-size chromosomes (200–900 kb) and approximately 100 mini-chromosomes (30–150 kb) (Wickstead et al., Reference Wickstead, Ersfeld and Gull2004; Daniels et al., Reference Daniels, Gull and Wickstead2010). The inheritance of intermediate-size- and mini-chromosomes is non-Mendelian, as they show mixed ploidy for analysed genetic markers (Alsford et al., Reference Alsford, Wickstead, Ersfeld and Gull2001). These chromosomes serve as depositories of genetic material used for the generation of novel variable surface glycoprotein (VSG) genes (Wickstead et al., Reference Wickstead, Ersfeld and Gull2004), which are instrumental for parasite survival and propagation in the mammalian bloodstream. As for Leishmania spp., their similar size genomes are split over 35–36 chromosomal pairs of smaller lengths (Myler, Reference Myler, Myler and Fasel2008; Cantacessi et al., Reference Cantacessi, Dantas-Torres, Nolan and Otranto2015).
The high gene density in trypanosomatids is explained by the near complete lack of introns and the relatively short intergenic regions (Günzl, Reference Günzl2010). Individual genes are arranged as same-strand tandem arrays that may include up to hundreds of genes. This organization is particularly pronounced in Leishmania, in which a megabase-sized chromosome may contain just two such clusters (Myler et al., Reference Myler, Audleman, de Vos, Hixson, Kiser, Lemley, Magness, Rickel, Sisk, Sunkin, Swartzell, Westlake, Bastien, Fu, Ivens and Stuart1999; Martínez-Calvillo et al., Reference Martínez-Calvillo, Nguyen, Stuart and Myler2004), whereas gene clusters in T. brucei are usually shorter (Bindereif, Reference Bindereif2012). Out of 191 transcription initiation sites were mapped in T. brucei, the majority (129) were found at the 5′ ends of the tandem clusters with the remaining sites localized within the clusters (Kolev et al., Reference Kolev, Franklin, Carmi, Shi, Michaeli and Tschudi2010). Unlike bacterial operons, trypanosomatid genes within the same cluster are not functionally related but seem to be arrayed randomly. However, the distance from the transcription initiation site within a cluster was crucial for the proper temporal expression of the heat shock and cell cycle-dependent genes. Moreover, such ‘positional bias’ was also observed for several functional gene groups, suggesting that temporal control by location within a cluster being an important principle of the T. brucei genome organization and expression (Kelly et al., Reference Kelly, Kramer, Schwede, Maini, Gull and Carrington2012).
Closely related species exhibit a remarkably high level of synteny in gene organization, and long regions of gene collinearity are observed even between distant relatives, such as between trypanosomes and leishmanias (Ghedin et al., Reference Ghedin, Bringaud, Peterson, Myler, Berriman, Ivens, Andersson, Bontempi, Eisen, Angiuoli, Wanless, Von Arx, Murphy, Lennard, Salzberg, Adams, White, Hall, Stuart, Fraser and El-Sayed2004; Peacock et al., Reference Peacock, Seeger, Harris, Murphy, Ruiz, Quail, Peters, Adlem, Tivey, Aslett, Kerhornou, Ivens, Fraser, Rajandream, Carver, Norbertczak, Chillingworth, Hance, Jagels, Moule, Ormond, Rutter, Squares, Whitehead, Rabbinowitsch, Arrowsmith, White, Thurston, Bringaud, Baldauf, Faulconbridge, Jeffares, Depledge, Oyola, Hilley, Brito, Tosi, Barrell, Cruz, Mottram, Smith and Berriman2007; Flegontov et al., Reference Flegontov, Butenko, Firsov, Kraeva, Eliáš, Field, Filatov, Flegontova, Gerasimov, Hlaváčová, Ishemgulova, Jackson, Kelly, Kostygov, Logacheva, Maslov, Opperdoes, O'Reilly, Sádlová, Ševčíková, Venkatesh, Vlček, Volf, Votýpka, Záhonová, Yurchenko and Lukeš2016). This conservation of gene order can be explained if spatial gene organization is implicated in the temporal control of gene expression, as it is in T. brucei. Nevertheless, group-specific differences were also documented. For example, while in all Trypanosoma spp. the arrays of rRNA genes comprising 28S, 18S and 5.8S rRNAs are well conserved and are repeated throughout the genome extending across several chromosomes to facilitate their high expression, in Leishmania spp. they are arranged as a single tandem array. The snRNAs genes occur within tRNA clusters in all trypanosomatids, although the location of these clusters varies among the species (Ivens et al., Reference Ivens, Peacock, Worthey, Murphy, Aggarwal, Berriman, Sisk, Rajandream, Adlem, Aert, Anupama, Apostolou, Attipoe, Bason, Bauser, Beck, Beverley, Bianchettin, Borzym, Bothe, Bruschi, Collins, Cadag, Ciarloni, Clayton, Coulson, Cronin, Cruz, Davies, De Gaudenzi, Dobson, Duesterhoeft, Fazelina, Fosker, Frasch, Fraser, Fuchs, Gabel, Goble, Goffeau, Harris, Hertz-Fowler, Hilbert, Horn, Huang, Klages, Knights, Kube, Larke, Litvin, Lord, Louie, Marra, Masuy, Matthews, Michaeli, Mottram, Muller-Auer, Munden, Nelson, Norbertczak, Oliver, O’Neil, Pentony, Pohl, Price, Purnelle, Quail, Rabbinowitsch, Reinhardt, Rieger, Rinta, Robben, Robertson, Ruiz, Rutter, Saunders, Schafer, Schein, Schwartz, Seeger, Seyler, Sharp, Shin, Sivam, Squares, Squares, Tosato, Vogt, Volckaert, Wambutt, Warren, Wedler, Woodward, Zhou, Zimmermann, Smith, Blackwell, Stuart, Barrell and Myler2005).
RNA polymerases and transcription
The tight spacing of protein-coding genes within clusters indicates the lack of individual promoters and the ability for independent gene transcription. Instead, it appears that RNA polymerase II (Pol II) initiates transcription at the ‘switch’ regions between the clusters or even randomly transcribes an entire cluster with a constant rate as a single polycistronic unit (Puechberty et al., Reference Puechberty, Blaineau, Meghamla, Crobu, Pages and Bastien2007; Das et al., Reference Das, Banday and Bellofatto2008; Kolev et al., Reference Kolev, Franklin, Carmi, Shi, Michaeli and Tschudi2010). The nascent polygenic RNA is processed co-transcriptionally. However, neither the promoters nor the transcription termination sites have been identified so far (Günzl et al., Reference Günzl, Vanhamme, Myler, Barry, McCulloch, Mottram and Acosta-Serrano2007; Myler, Reference Myler, Myler and Fasel2008). The only well-characterized Pol II promoters are those for transcription of spliced leader (SL) RNA genes (Gilinger and Bellofatto, Reference Gilinger and Bellofatto2001; Dossin Fde and Schenkman, Reference Dossin Fde and Schenkman2005). These small non-coding transcripts are used during mRNA maturation, hence hundreds of individual SL RNA genes are present in the trypanosomatid genome in order to sustain the necessary rate of mRNA processing (Liang et al., Reference Liang, Haritan, Uliel and Michaeli2003; Lee et al., Reference Lee, Nguyen, Schimanski and Gunzl2007b). These genes are arranged as clusters of tandem units, but each gene is individually transcribed by Pol II using a promoter and a transcription termination signal. Pol II itself is composed of 12 rather conservative subunits (Das et al., Reference Das, Li, Liu and Bellofatto2006; Martinez-Calvillo et al., Reference Martinez-Calvillo, Saxena, Green, Leland and Myler2007). Its unique property is the absence of the conserved heptad amino acid sequence in the carboxy-terminal domain (CTD) of the largest subunit RPB1. This difference apparently reflects the fact that a co-transcriptional capping of a monocistronic pre-mRNA (mediated by CTD in other organisms) does not take place in trypanosomatids (see below). The pre-initiation complex assembled at the SL RNA promoter includes recognizable homologues of metazoan basal transcription factors, such as TRF4 (TATA-box binding protein-related factor 4) and some subunits of TFIIH (Ivens et al., Reference Ivens, Peacock, Worthey, Murphy, Aggarwal, Berriman, Sisk, Rajandream, Adlem, Aert, Anupama, Apostolou, Attipoe, Bason, Bauser, Beck, Beverley, Bianchettin, Borzym, Bothe, Bruschi, Collins, Cadag, Ciarloni, Clayton, Coulson, Cronin, Cruz, Davies, De Gaudenzi, Dobson, Duesterhoeft, Fazelina, Fosker, Frasch, Fraser, Fuchs, Gabel, Goble, Goffeau, Harris, Hertz-Fowler, Hilbert, Horn, Huang, Klages, Knights, Kube, Larke, Litvin, Lord, Louie, Marra, Masuy, Matthews, Michaeli, Mottram, Muller-Auer, Munden, Nelson, Norbertczak, Oliver, O’Neil, Pentony, Pohl, Price, Purnelle, Quail, Rabbinowitsch, Reinhardt, Rieger, Rinta, Robben, Robertson, Ruiz, Rutter, Saunders, Schafer, Schein, Schwartz, Seeger, Seyler, Sharp, Shin, Sivam, Squares, Squares, Tosato, Vogt, Volckaert, Wambutt, Warren, Wedler, Woodward, Zhou, Zimmermann, Smith, Blackwell, Stuart, Barrell and Myler2005). Biochemical analyses revealed additional rather divergent subunits of TFIIH, as well as TFIIA, TFIIB and Mediator complex (Das and Bellofatto, Reference Das and Bellofatto2003; Schimanski et al., Reference Schimanski, Nguyen and Gunzl2005; Reference Schimanski, Brandenburg, Nguyen, Caimano and Gunzl2006; Lee et al., Reference Lee, Jung and Gunzl2009; Reference Lee, Cai, Panigrahi, Dunham-Ems, Nguyen, Radolf, Asturias and Gunzl2010), to the total of more than 20 proteins.
The peculiar utilization of RNA polymerases in trypanosomatids is further illustrated by the participation of Pol I in the transcription of the protein-coding genes. This enzyme, canonically serving to transcribe ribosomal RNA genes, also performs that function in trypanosomatids (Hernandez and Cevallos, Reference Hernandez and Cevallos2014). However, in T. brucei it also transcribes a special group of genes, which are located in the subtelomeric regions of large chromosomes, namely the expressed version of the VSG genes and a group of expression site-associated genes (ESAGs) (Vanhamme and Pays, Reference Vanhamme and Pays1995; Navarro and Gull, Reference Navarro and Gull2001; Günzl et al., Reference Günzl, Bruderer, Laufer, Schimanski, Tu, Chung, Lee and Lee2003). Another class of protein-coding genes transcribed by Pol I in T. brucei is procyclin, which constitutes the major surface component of the procyclic stage (Günzl et al., Reference Günzl, Bruderer, Laufer, Schimanski, Tu, Chung, Lee and Lee2003). The transcriptionally active Pol I complex contains at least 12 subunits, most of which are conserved but at least one is trypanosomatid-specific (Nguyen et al., Reference Nguyen, Schimanski and Gunzl2007). Each large chromosome has two VSG Pol I promoters in its subtelomeric regions. These promoters are composed of two short sequence element and are structurally different from the ribosomal RNA and procyclin promoters with a more complex architecture, however, their recognition depends on the same multi-subunit transcription factor CITFA (Brandenburg et al., Reference Brandenburg, Schimanski, Nogoceke, Nguyen, Padovan, Chait, Cross and Gunzl2007; Kolev et al., Reference Kolev, Gunzl and Tschudi2017). There is a total ~ 20 subtelomeric expression sites (ES) for VSG genes (~5 ES's for metacyclic VSG genes and ~15 ES's for bloodstream VSG genes), but in a given cell only one ES is active at any time (Navarro et al., Reference Navarro, Cross and Wirtz1999). A bloodstream ES is 45–60 kbp long and includes 9–10 ESAGs in addition to a single telomere-proximal VSG gene separated from the upstream ESAGs by a long block of 70 bp repeats. A metacyclic ES is short (up to 6 kb), lacks ESAGs and a repeat block. The ESAG and VSG genes in the active site are transcribed by in form of a polycistronic unit, with the RNA processing occurring co-transcriptionally. The choice of the single active ES is regulated epigenetically (Günzl et al., Reference Günzl, Kirkham, Nguyen, Badjatia and Park2015; Maree et al., Reference Maree, Povelones, Clark, Rudenko and Patterton2017). Transcription of all the inactive ESs also gets initiated, but terminates prematurely before reaching the promoter-distant VSG gene due to telomere-dependent epigenetic silencing (Batram et al., Reference Batram, Jones, Janzen, Markert and Engstler2014; Kassem et al., Reference Kassem, Pays and Vanhamme2014). This silencing is, at least in part, mediated by a telomeric protein RAP1, whose depletion results in de-repression of the silent ESs (Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009). So does depletion of the histone H3 methylase DOT1 indicating that chromatin structure also plays important role in this silencing (Figueiredo et al., Reference Figueiredo, Janzen and Cross2008). However, expression level at the derepressed ESs does not achieve that of the active ES, indicating that additional controls are at play, including transcription initiation (Nguyen et al., Reference Nguyen, Muller, Park, Siegel and Gunzl2014; Günzl et al., Reference Günzl, Kirkham, Nguyen, Badjatia and Park2015). A possible mechanism can be based sequestration of all necessary factors in a single transcription focus. This sequestration can be enforced by a recently discovered VEX1 (VSG exclusion) factor (Glover et al., Reference Glover, Hutchinson, Alsford and Horn2016). Localized in a single nuclear focus next to ESB, VEX1 is proposed to exert both negative regulation (on silent ESs) and positive regulation (on the active ES). Its molecular mechanism is unclear but it appears to be homology-based. Stage-specific transcription of the epimastigote coat protein BARP (brucei alanine-rich protein) may also be Pol I-dependent (Urwyler et al., Reference Urwyler, Studer, Renggli and Roditi2007; Savage et al., Reference Savage, Kolev, Franklin, Vigneron, Aksoy and Tschudi2016).
Finally, RNA polymerase III (Pol III) is the only polymerase in trypanosomes that retained its canonical functions, transcribing the tRNA and 5S rRNA genes (Das et al., Reference Das, Banday and Bellofatto2008).
mRNA processing by trans-splicing coupled with 3′-end cleavage and polyadenylation
All mature mRNAs in trypanosomatids contain a non-coding 39 nucleotide-long SL RNA (Parsons et al., Reference Parsons, Nelson, Watkins and Agabian1984). These add-on sequences are derived from the initial SL RNA gene transcripts, which in addition to the mini-exon on the 5′-end contain a variable species-dependent length mini-intron on the 3′-end (Goncharov et al., Reference Goncharov, Xu, Zimmer, Sherman and Michaeli1998; Mandelboim et al., Reference Mandelboim, Estrano, Tschudi, Ullu and Michaeli2002). There is a co-transcriptionally added hyper-methylated cap 4. This structure contains m7GpppG cap on the 5′-end and 2′-O methylations at nucleotides 1 and 2, commonly seen in other organisms (Perry et al., Reference Perry, Watkins and Agabian1987). Unique to trypanosomatids, it also contains 2′-O methyl groups at nucleotides 3 and 4, and methylated bases at nucleotides 1 (m26A) and 4 (m3U) (Ullu and Tschudi, Reference Ullu and Tschudi1993). The functional significance of the trypanosomatid-specific hyper-methylated cap remains unclear (Sturm et al., Reference Sturm, Zamudio, Campbell and Bindereif2012).
The capped SL is added to mRNA by trans-splicing and is essential for stability and translatability of the latter (Sturm et al., Reference Sturm, Zamudio, Campbell and Bindereif2012). Although trans-splicing is not unique to trypanosomatids, and occurs, along with conventional cis-splicing, in some Metazoa and protists (Lukeš et al., Reference Lukeš, Leander and Keeling2009), in trypanosomatids it is a major and obligatory step in the maturation of each mRNA. In addition, due to continuous transcription of protein-coding gene clusters, the processes of trans-splicing and 3′-end cleavage/polyadenylation are tightly coupled (LeBowitz et al., Reference LeBowitz, Smith, Rusche and Beverley1993; Matthews et al., Reference Matthews, Tschudi and Ullu1994). Intergenic regions included in the nascent RNA contain the cleavage/polyadenylation sites positioned 3′ to each coding region. Cleavage of the nascent RNA at this site not only enables the 3′ maturation of the upstream pre-mRNA but also liberates the 5′-end of the downstream pre-mRNA for participation in trans-splicing. Mechanistically, the process of trans-splicing includes two trans-esterification reactions as in conventional cis-splicing (Günzl, Reference Günzl2010). The SL RNA participates in the reaction as a specific snRNP particle (Goncharov et al., Reference Goncharov, Palfi, Bindereif and Michaeli1999), apparently substituting the U1 snRNP in a trans-spliceosome. Other snRNPs, U2, U4–U6, have also been identified in trypanosomatids (Palfi et al., Reference Palfi, Xu and Bindereif1994). Interestingly, the U1 snRNP, typically involved in 5′ splice site recognition is also present, because at least two T. brucei genes – poly(A)-polymerase and DEAD/H RNA helicase – contain a cis-splicing intron (Mair et al., Reference Mair, Shi, Li, Djikeng, Aviles, Bishop, Falcone, Gavrilescu, Montgomery, Santori, Stern, Wang, Ullu and Tschudi2000; Siegel et al., Reference Siegel, Hekstra, Wang, Dewell and Cross2010).
The absence of individual promoters and the constant rate of transcription by Pol II of most protein-coding genes dictate that gene regulation does not operate at the level of transcription initiation, in contrast to most other eukaryotes. Instead, gene expression is mainly controlled post-transcriptionally, with the main level being mRNA stability (McNicoll et al., Reference McNicoll, Muller, Cloutier, Boilard, Rochette, Dube and Papadopoulou2005; Requena, Reference Requena2011). The abundance of mRNA depends on its half-life, which averages around 20 min in this life stage (Fadda et al., Reference Fadda, Ryten, Droll, Rojas, Farber, Haanstra, Merce, Bakker, Matthews and Clayton2014; Kramer, Reference Kramer2017b). A mature mRNA is 5′-capped and 3′-polyadenylated and its exonucleolytic degradation by the exosome requires removal of either structural feature.
Stability of mRNA is mainly defined by the structure of its 3′ untranslated region (3′-UTR). Typically, it is long enough (~400 nt) to accommodate several RNA-binding proteins (Nozaki and Cross, Reference Nozaki and Cross1995; De Gaudenzi et al., Reference De Gaudenzi, Frasch and Clayton2005), of which there is a diverse population with varying functions, binding constants and copy numbers per cell (Erben et al., Reference Erben, Fadda, Lueong, Hoheisel and Clayton2014; Lueong et al., Reference Lueong, Merce, Fischer, Hoheisel and Erben2016). Some of those proteins facilitate mRNA degradation by recruiting deadenylating or decapping factors (Kramer, Reference Kramer2017a), or exosomes (Fadda et al., Reference Fadda, Farber, Droll and Clayton2013), while others stabilize the mRNA either directly by protecting it from degradation or indirectly by competing with the degradation factors (Estevez, Reference Estevez2008). These proteins are engaged in dynamic interactions with the mRNA and define the longevity of a given transcript.
Multiplicity of translation factors
Additional regulation occurs at the translation level, as demonstrated by the abundance of a given protein frequently not correlating with levels of its encoding mRNA (McNicoll et al., Reference McNicoll, Drummelsmith, Muller, Madore, Boilard, Ouellette and Papadopoulou2006; Leifso et al., Reference Leifso, Cohen-Freue, Dogra, Murray and McMaster2007). In other eukaryotes, translation is often regulated at the stage of initiation and this also seems to be the case in trypanosomatids (Rezende et al., Reference Rezende, Assis, Nunes, da Costa Lima, Marchini, Freire, Reis and de Melo Neto2014). However, as indicated by the uniquely complex 5′-end structure of mRNA, in trypanosomatids this process has deviated from the canonical eukaryotic pattern. At least four paralogs of eIF4E and six of eIF4 G were identified by genome analysis (Zinoviev and Shapira, Reference Zinoviev and Shapira2012). While their functions are not fully ascribed, the available evidence indicates that trypanosomatid eIF4E-1 and eIF4E-4 are the bona fide parts of the respective eIF4F complexes and may be involved in life cycle stage-specific (developmental) regulation of translation (Yoffe et al., Reference Yoffe, Leger, Zinoviev, Zuberek, Darzynkiewicz, Wagner and Shapira2009; Zinoviev et al., Reference Zinoviev, Leger, Wagner and Shapira2011), while eIF4E-2 may mediate mRNA–ribosome interactions during elongation (Yoffe et al., Reference Yoffe, Zuberek, Lewdorowicz, Zeira, Keasar, Orr-Dahan, Jankowska-Anyszka, Stepinski, Darzynkiewicz and Shapira2004). Unlike in higher eukaryotes, where eIF4G mediates interactions between eIF4E and 3′-bound poly-A binding protein (PABP), in trypanosomatids this interaction is performed directly by eIF4E (−1 or −4), while the subunit eIF4G-3 links the former with eIF4A1, which is assumed to be involved in recognition of the initiation codon (Pestova et al., Reference Pestova, Kolupaeva, Lomakin, Pilipenko, Shatsky, Agol and Hellen2001). Out of the three isoforms of PABP in trypanosomatids, only PABP-1 participates in the formation of the cap-dependent initiation complex (Kramer et al., Reference Kramer, Bannerman-Chukualim, Ellis, Boulden, Kelly, Field and Carrington2013).
Trypanosomatids lack the homologue of a small eIF4E-binding protein (4E-BP), which in higher eukaryotes can block the formation of the cap-bound initiation complex, and hence translation, by preventing the interaction between the eIF4E and eIF4G subunits. Instead, trypanosomatids possess a different type of 4E-BP, called 4E-IP, which interacts with eIF4E-1 (Zinoviev et al., Reference Zinoviev, Leger, Wagner and Shapira2011). In Leishmania, this protein appears to participate in stage-specific phosphorylation-dependent translation control (Zinoviev and Shapira, Reference Zinoviev and Shapira2012).
Unexpected codon reassignment
The already long list of oddities encountered in trypanosomatids has been recently extended by a unique codon reassignment found in several representatives of the genus Blastocrithidia. Here, all three stop codons are reassigned to code for amino acids (Záhonová et al., Reference Záhonová, Kostygov, Ševčíková, Yurchenko and Eliáš2016), a deviation paralleled only by two ciliate species (Swart et al., Reference Swart, Serra, Petroni and Nowacki2016). Most changes of the genetic code involve reassignment of stop codon(s), in particular, UGA to decode Trp in many mitochondria and bacteria, yet almost always at least one stop codon is retained for terminating translation (Keeling, Reference Keeling2016). Trypanosomatids are no exception and use UAG to specify Trp in their kinetoplast DNA, which does not encode any tRNA genes (de la Cruz et al., Reference de la Cruz, Neckelmann and Simpson1984). Consequently, all tRNAs have to be imported into the mitochondrion from the cytosol (Simpson et al., Reference Simpson, Suyama, Dewes, Campbell and Simpson1989). Since there is only a single tRNATrp in the trypanosomatid nuclear genome, the anticodon of which recognizes the standard Trp UGG codon, in order to decode UGA the tRNATrp undergoes C to U editing at the first position of its anticodon. However, to prevent read-through of the UGA stop codon in the cytoplasm, the deamination must happen only after import into the organelle (Alfonzo et al., Reference Alfonzo, Blanc, Estevez, Rubio and Simpson1999). While the enzyme responsible for this single-site editing has yet to be identified, trypanosomatids were shown to possess two distinct tryptophanyl-tRNA synthetases to charge tRNATrp in the mitochondrion and the cytosol (Charriere et al., Reference Charriere, Helgadottir, Horn, Soll and Schneider2006). In Blastocrithidia spp., however, codon reassignment happens also in the cytosol, as UAR and UGA code for Glu and Trp, respectively, with UAA being also used as a genuine stop codon. It remains to be established how these flagellates distinguish between in-frame and genuine stops so that proper translation termination can occur.
Kinetoplast, kinetoplast DNA and RNA editing
These aspects represent one of the major trypanosomatid ‘oddities’ justifying the attention, including a historical coverage, given to this subject.
Organization of the kinetoplast DNA
The defining feature of the class Kinetoplastea is the existence of the kinetoplast, a particular region of the cell's single mitochondrion, with the bulk of mitochondrial DNA. Due to its intense staining with the basophilic dyes, this structure could be easily detected using light microscopy, which facilitated its discovery more than a century ago (Laveran and Mesnil, Reference Laveran and Mesnil1901). The kinetoplast's location adjacent to the basal body of the flagellum led early researchers to believe that this organelle might be involved in the flagellar kinetic properties and named it accordingly (Alexeieff, Reference Alexeieff1917). With the advent of electron microscopy, it was found that kinetoplast contains highly compacted DNA (termed kinetoplast DNA or kDNA) and that purified kDNA represents a network composed of thousands of catenated heterogeneous minicircles (Steinert, Reference Steinert1960; Kleisen and Borst, Reference Kleisen and Borst1975; Vickerman, Reference Vickerman, Vickerman and Preston1976). The size of minicircles was species-specific and in most cases varies from 1 to 2.5 kb, although species with larger size minicircles were also found (Kidane et al., Reference Kidane, Hughes and Simpson1984; Yurchenko et al., Reference Yurchenko, Hobza, Benada and Lukeš1999). Each minicircle molecule contains from one to four (depending on the species) conserved regions with the remaining sequence forming the variable region(s) (Ray, Reference Ray1989; Simpson, Reference Simpson1997; Yurchenko and Kolesnikov, Reference Yurchenko and Kolesnikov2001). The number of sequence classes can differ greatly even among the related species, with some species (e.g. T. equiperdum) having almost homogeneous minicircles and others (e.g. T. brucei) displaying hundreds of classes of minicircles per network (Lai et al., Reference Lai, Hashimi, Lun, Ayala and Lukeš2008; Koslowsky et al., Reference Koslowsky, Sun, Hindenach, Theisen and Lucas2014). These properties of minicircles, as well as the localization of the protein-coding genes, remained enigmatic until the discovery of maxicircles and RNA editing. The former was detected as a minor component of kDNA with the size (varying from 20 kb in T. brucei to 40 kb in T. cruzi) comparable with that of other mitochondrial genomes (Borst and Fase-Fowler, Reference Borst and Fase-Fowler1979; Simpson, Reference Simpson1979). Maxicircles from all investigated species contained a 16 kb ‘conserved’ region with the colinear arrangement of the cross-hybridizing DNA fragments (Muhich et al., Reference Muhich, Simpson and Simpson1983; Maslov et al., Reference Maslov, Kolesnikov and Zaitseva1984). Maxicircle size differences were attributed to a ‘divergent’ region that is composed of repeats highly variable in size and sequence (Muhich et al., Reference Muhich, Neckelmann and Simpson1985; Horváth et al., Reference Horváth, Maslov, Peters, Haviernik, Wuestenhagen and Kolesnikov1990; Flegontov et al., Reference Flegontov, Strelkova and Kolesnikov2006). This region may contain the origins of maxicircle DNA replication (Myler et al., Reference Myler, Glick, Feagin, Morales and Stuart1993), but its exact function remains elusive even today.
Maxicircles and RNA editing
DNA sequencing of the conserved region in L. tarentolae and T. brucei revealed a set of protein-coding genes typical for mitochondria: 12S (large subunit) and 9S (small subunit) ribosomal RNA genes, three subunits of cytochrome c oxidase (COI, COII and COIII), a subunit (Cyb) of the cytochrome bc 1 complex, several subunits of NADH dehydrogenase (ND1, ND3, ND5, ND7) and one subunit of F1Fo ATP synthase (A6) (Benne et al., Reference Benne, De Vries, Van den Burg and Klaver1983; de la Cruz et al., Reference de la Cruz, Neckelmann and Simpson1984). There were also six G-rich regions (G1–G6) and several reading frames with no recognizable function (MURF1, MURF2, MUR5) (Simpson et al., Reference Simpson, Douglass, Lake, Pellegrini and Li2015). Surprisingly, some of the identified genes appeared to be defective: thus, the proper initiator codons were absent in COIII and Cyb, and there was a −1 frameshift in COII. Analysis of the cDNA in C. fasciculata showed that the −1 frameshift in the COII DNA sequence is ‘edited’ by insertion of four U-residues in the mRNA (Benne et al., Reference Benne, Van den Burg, Brakenhoff, Sloof, Van Boom and Tromp1986). Subsequently, it was shown that RNA editing is responsible for repairing the aforementioned defects present in the original (pre-edited) mRNAs thereby converting the pre-edited transcripts into translatable (fully edited) mRNAs (van der Spek et al., Reference van der Spek, van den Burg, Croiset, van den Broek, Sloof and Benne1988; Feagin et al., Reference Feagin, Shaw, Simpson and Stuart1988b). The amount of editing required for different transcripts varies drastically. Thus, a relatively modest editing by insertion of a dozen or so (and removal of a smaller number) of U-residues takes place in Cyb, MURF2, ND7 and COIII mRNAs in L. tarentolae (5′-editing and internal frameshift correction). On the other side of the spectrum are the A6, COIII and ND7 mRNAs of T. brucei, which emerge from pre-edited transcripts by incorporating hundreds of U-residues (and also deleting a small number of some of the maxicircle-encoded U-residues) (Feagin et al., Reference Feagin, Abraham and Stuart1988a; Koslowsky et al., Reference Koslowsky, Bhat, Perrollaz, Feagin and Stuart1990). Such cases of massive editing were termed ‘pan-editing’, while the respective genomic regions were termed ‘cryptogenes’. In addition, six maxicircle G-rich regions turned out to represent pan-edited cryptogenes for five NADH dehydrogenase subunits and ribosomal protein S12 (RPS12) (Maslov et al., Reference Maslov, Sturm, Niner, Gruszynski, Peris and Simpson1992; Read et al., Reference Read, Myler and Stuart1992; Thiemann et al., Reference Thiemann, Maslov and Simpson1994). The editing of A6 transcript extends its reading frame by almost one third of its original length in L. tarentolae (Maslov and Simpson, Reference Maslov and Simpson1992), and it is pan-edited in T. brucei (Bhat et al., Reference Bhat, Koslowsky, Feagin, Smiley and Stuart1990). Thus, the maxicircles in both flagellates contain the same set of genes, but vary in the amount of editing for some of them. The other studied trypanosomatids have the same gene organization pattern, with the notable exception of the plant parasites Phytomonas spp., which lack cytochrome c oxidase and apocytochrome b complexes in its inner mitochondrial membrane (Maslov et al., Reference Maslov, Nawathean and Scheel1999; Nawathean and Maslov, Reference Nawathean and Maslov2000). Accordingly, genes for the respective subunits (COI–COIII and Cyb) are missing from the maxicircle conserved region, while the rest of its gene content remains intact.
The question regarding the source of the sequence information for guiding was resolved by the search for maxicircle sequences complementary (allowing G-U base pairing) to the fully edited sequences of COII, ND7, MURF2 and Cyb. This analysis led to the identification of small transcripts, termed guide (g) RNAs (Blum et al., Reference Blum, Bakalara and Simpson1990). Soon thereafter, gRNA genes were discovered in the variable region of minicircles, solving the long-standing mystery of the functional role of these molecules (Pollard et al., Reference Pollard, Rohrer, Michelotti, Hancock and Hajduk1990; Sturm and Simpson, Reference Sturm and Simpson1990a). The rationale for partitioning the gRNA genes between the maxicircles and the minicircles remains unclear, as in some cases both types participate in the editing of the same transcript. The mature gRNAs are 40–50 nt long and contain a post-transcriptionally added oligo(U)-tail on the 3′-end (Blum and Simpson, Reference Blum and Simpson1990). As the result of editing, a perfect sequence match is achieved between the mRNA and gRNA sequences. A single gRNA is sufficiently long to cover a stretch of the pre-edited sequence, which typically includes less than 20–30 insertions and a few deletions, termed ‘editing block’ (Simpson et al., Reference Simpson, Maslov, Blum and Benne1993). Cryptogene-derived mRNAs are edited over the entire length and require editing by multiple gRNAs. The editing begins at the 3′-end of a pre-edited transcript and gradually spreads upstream so that the 5′-end of the mRNA is edited last (Sturm and Simpson, Reference Sturm and Simpson1990b; Maslov et al., Reference Maslov, Sturm, Niner, Gruszynski, Peris and Simpson1992).
When there is little or no redundancy in the gRNA content, a loss of a single gRNA would render completion of editing impossible. The stochastic nature of minicircle inheritance during the cell division makes such a loss a real possibility (Savill and Higgs, Reference Savill and Higgs1999). The ensuing disruption of the productive editing for even a single gene is likely to be lethal when each of the maxicircle products is required at least at some stage of the parasite's life cycle. Thus, the selection ensures the maintenance of a full editing capability in natural populations. However, mutants with editing loss for a dispensable product can survive in culture. This is the case of some strains of L. tarentolae, which display disrupted editing of several pan-edited mRNAs due to the loss of minicircle classes (Thiemann et al., Reference Thiemann, Maslov and Simpson1994). A similar disruption of editing was observed for several strains of C. fasciculata, L. donovani and P. serpens (Sloof et al., Reference Sloof, Arts, van den Burg, van der Spek and Benne1994; Maslov et al., Reference Maslov, Hollar, Haghighat and Nawathean1998; Neboháčová et al., Reference Neboháčová, Kim, Simpson and Maslov2009). So far, the only studied species, which maintain the full editing capacity in culture are L. mexicana (Maslov, Reference Maslov2010) and T. brucei, the latter case likely due to the high redundancy of its gRNA repertoire (Corell et al., Reference Corell, Feagin, Riley, Strickland, Guderian, Myler and Stuart1993; Riley et al., Reference Riley, Corell and Stuart1994). It should be mentioned that in T. brucei, unlike other species, each minicircle encodes up to three different gRNAs and the number of minicircle classes is comparatively high, suggesting that this species is more refractory to an occasional loss of a minicircle class (Hong and Simpson, Reference Hong and Simpson2003).
Alternatively and partially edited RNA molecules may co-exist together with the fully edited mRNAs, contributing to the diversity of mitochondrial proteins (Ochsenreiter et al., Reference Ochsenreiter, Cipriano and Hajduk2008; Hajduk and Ochsenreiter, Reference Hajduk and Ochsenreiter2010; Gerasimov et al., Reference Gerasimov, Gasparyan, Kaurov, Tichy, Logacheva, Kolesnikov, Lukeš, Yurchenko, Zimmer and Flegontov2018).
Evolution of editing
The evolutionary origin of editing and the rationale for its existence remain obscure (Simpson and Maslov, Reference Simpson and Maslov1994a; Lukeš et al., Reference Lukeš, Hashimi and Zíková2005). So far, there is no satisfactory scenario explaining the origin of this process from the metabolic or gene regulation standpoint regardless of whether or not it was subsequently employed for any such purpose. An attractive hypothesis is the origin by constructive neutral evolution (CNE) (Lukeš et al., Reference Lukeš, Archibald, Keeling, Doolittle and Gray2011; Gray, Reference Gray2012), yet this remains a speculative scenario. CNE is a neutral evolutionary theory which aims to explain some aspects of cellular complexity by mechanisms that do not rely on positive selection (Stoltzfus, Reference Stoltzfus1999). In this scenario, numerous T-deletions or insertions in kDNA would be tolerated due to the fortuitous interactions made possible by the pre-existence of enzymatic activities capable of restoring these mutations at the RNA levels (‘presuppression’). Such activities, e.g. endo- and exonucleases, RNA ligase, would be derived from the cellular systems originally serving some other purpose(s). However, such interactions would lead to eventual formation of the dependence on such activities for kDNA gene expression, and thus, to preservation and further evolution of the editing machinery. A somewhat extended version called ‘irremediable complexity’ postulates that when a given cellular component acquires mutations making it dependent on another component, such dependence will become complex and irreversible (Lukeš et al., Reference Lukeš, Archibald, Keeling, Doolittle and Gray2011). Thus, CNE is evolutionary ratchet responsible for a steady increase of overall organismal complexity. However, selective advantages were also associated with the emergence of editing. One scenario postulates that as a consequence of pan-editing, information necessary for production of several proteins is spread over the kDNA, preventing loss of genes in parts of the life cycle when their products are not required (Speijer, Reference Speijer2006). In any case, since the U-insertion/deletion type of editing is also found in various bodonids (Lukeš et al., Reference Lukeš, Arts, van den Burg, de Haan, Opperdoes, Sloof and Benne1994; Maslov and Simpson, Reference Maslov and Simpson1994; Blom et al., Reference Blom, de Haan, van den Berg, Sloof, Jirku, Lukeš and Benne1998), while it is absent from their sister group Diplonemida (Faktorová et al., Reference Faktorová, Valach, Kaur, Burger, Lukeš, Gray and Cruz-Reyes2018), its origin likely coincided with that of the entire taxon Kinetoplastea, and for that matter with the origin of the kinetoplast itself (Lukeš et al., Reference Lukeš, Guilbride, Votýpka, Zíková, Benne and Englund2002; Simpson et al., Reference Simpson, Lukeš and Roger2002). The kDNA essentially represents the depository for the gRNA genes, so it is likely that its various forms emerged as different evolutionary answers to the problem of how to organize and maintain the extensive gRNA diversity in proximity to the editing itself (Lukeš et al., Reference Lukeš, Guilbride, Votýpka, Zíková, Benne and Englund2002). A minicircle-based concatenated network is the type that appeared in the ancestral trypanosomatids, and it is likely that pan-editing is also an ancestral trait for this taxon. If we assume, that editing per se does not play a significant or vital role, but is merely a product of the CNE, then it represents a substantial burden for the cells. However, the cells depend on it for mitochondrial mRNA production and, unlike in culture, a loss of productive editing in nature would be lethal due to the parasite's inability to complete its life cycle. The gRNA redundancy and diversity observed in T. brucei may have been developed in this phylogenetic lineage for preservation of the editing in spite of an occasional minicircle loss. A different evolutionary trend is observed in other trypanosomatid lineages, such as Leishmania and some monoxenous species, in which the ancestral cryptogenes appear to have been substituted by their less-edited counterparts (Maslov et al., Reference Maslov, Avila, Lake and Simpson1994; Simpson and Maslov, Reference Simpson and Maslov1994a). The replacement might have occurred via homologous recombination between a cDNA copy of the partially edited mRNA and the cryptogene (Simpson and Maslov, Reference Simpson and Maslov1994b). This would in turn eliminate the essentiality of several gRNAs and the respective minicircle classes. As the copy numbers of the remaining minicircles would proportionally increase, the likelihood of their loss due to mis-segregation would decrease, thereby creating a more stable genetic system in the kinetoplast. The only cryptogene apparently unaffected by the replacement trend is RPS12, which is invariably pan-edited in all studied trypanosomatid and bodonid species. This may be related to the fact that this mRNA encodes an indispensable mitoribosomal protein and any change in its synthesis would impact translation of all mitochondrial transcripts, including those, which do not require editing, e.g. COI. Thus, preserving pan-editing of RPS12 mRNA might be important for coordination of the editing and translation systems (Aphasizheva et al., Reference Aphasizheva, Maslov and Aphasizhev2013).
Kinetoplast DNA replication
The problem of minicircle loss is alleviated to some degree by the evolution of a unique mechanism of kDNA replication, described here for T. brucei and other trypanosomatids, all of which have a catenated network composed of relaxed circles. Synthesis of kDNA occurs during the S phase of the cell cycle, while segregation of the daughter networks, along with the tightly coupled process of the flagellar duplication, is completed during the G2 phase (Simpson and Kretzer, Reference Simpson and Kretzer1997). This strict timing is controlled by a cell cycle-dependent regulation of the key enzymes participating in the process (Hines and Ray, Reference Hines and Ray1997; Li et al., Reference Li, Sun, Hines and Ray2007). The replication process has been described in a series of recent reviews (Klingbeil and Englund, Reference Klingbeil and Englund2004; Liu et al., Reference Liu, Liu, Motyka, Agbo and Englund2005; Jensen and Englund, Reference Jensen and Englund2012; Povelones, Reference Povelones2014), and is presented here only briefly. The minicircles are released from the covalently closed network and replicated in the kinetoflagellar zone (KFZ), which represents an intra-mitochondrial compartment between the kDNA and the basal body of the flagellum. The nicked or gapped daughter molecules are reattached to the network's periphery at the two antipodal sites, thereby slowly increasing the network's size. A yet unknown mechanism rotates the replicating network to ensure an even distribution of the reattached molecules. When all minicircles have been replicated, the kDNA network doubles in size and each minicircle contains nicks, which are closed before the network splits into two. This apparently highly complex mechanism serves to ascertain that each minicircle replicates only once. The antipodal attachment and network rotation likely serve to maximize the chances for the two daughter minicircles to segregate into the different networks during network division. Throughout the cell cycle, the kDNA network remains associated with the flagellar basal and parabasal bodies by a filamentous structure, termed TAC (tripartite attachment complex). This physical connection is thought to ascertain the coordinated duplication of the kDNA network and the flagellar apparatus.
During late stages of the kDNA replication, the two segregating sister kDNA networks remain for some time attached by a thin yet morphologically prominent connector, termed umbiliculum or nabelschnur, a filamentous structure which is cut at the final stage of the daughter network segregation (Gluenz et al., Reference Gluenz, Shaw and Gull2007). It is likely composed of numerous dedicated proteins, with leucine aminopeptidase 1 being the only one identified so far (Pena-Diaz et al., Reference Pena-Diaz, Vancova, Resl, Field and Lukeš2017).
Core catalytic activities of RNA editing
The recapitulation of the U-insertion or U-deletion at a single editing site (ES) in vitro using synthesized double-stranded (ds) RNA substrates and mitochondrial lysates supported the original hypothesis that trypanosome RNA editing requires a cascade of enzymatic activities (Blum et al., Reference Blum, Bakalara and Simpson1990; Kable et al., Reference Kable, Seiwert, Heidmann and Stuart1996; Seiwert et al., Reference Seiwert, Heidmann and Stuart1996). These catalytic steps are orchestrated by the RNA editing core complex (RECC), also known as the 20S editosome or L-complex, the former alias reflecting the sedimentation coefficient of the enzymatically active complex (Simpson et al., Reference Simpson, Aphasizhev, Gao and Kang2004; Stuart et al., Reference Stuart, Schnaufer, Ernst and Panigrahi2005; Read et al., Reference Read, Lukeš and Hashimi2016). In T. brucei, RECC is made up of about 20 components abbreviated as kinetoplastid RNA editing (KRE) proteins (Stuart et al., Reference Stuart, Schnaufer, Ernst and Panigrahi2005). These subunits include two RNA ligases (KREL1 and 2) and U-specific exonucleases (KREX1 and 2), a terminal U transferase (KRET2) and three RNase III endonucleases (KREN1-3). Other subunits such as six OB-fold bearing proteins (KREPA1-6) serve a structural and/or RNA- binding role.
RECC catalyses the following steps. First, at the gRNA defined ES, the mRNA is cleaved by one of the KRENs to yield 5′ and 3′ fragments that are bridged by the bound gRNA. Next, depending on the gRNA information domain sequence, one or more Us are added by KRET2 or removed by KREX2 from the 5′-fragment. The catalytic activity of KREX1 is dispensable in vivo and the protein may play a more structural role (Rogers et al., Reference Rogers, Gao and Simpson2007; Carnes et al., Reference Carnes, Lewis Ernst, Wickham, Panicucci and Stuart2012). Finally, after the ES has been edited to be complementary to the gRNA information domain, the two mRNA fragments are sealed together by KREL1. The other ligase KREL2 plays a still undefined role in RNA editing that is expendable for this final step (Gao and Simpson, Reference Gao and Simpson2003).
The recognition that a given ES requires U-insertion, U-deletion or the editing of cis-gRNA-containing COX2 is mediated by one of the three endonucleases, which specifically cleave only one type of dsRNA substrate (Carnes et al., Reference Carnes, Trotter, Peltan, Fleck and Stuart2008). Typically, RNase III endonucleases form homodimers to cleave both dsRNA strands (MacRae and Doudna, Reference MacRae and Doudna2007). Because RNA editing requires only the cleavage of the mRNA strand, each KREN protein dimerizes with its own unique catalytically inert partner (Carnes et al., Reference Carnes, Soares, Wickham and Stuart2011). It remains unclear whether these KREN containing RECCs represent different, stable isoforms that specialize in processing a specific ES type, or if they represent discrete modules that are selectively added onto RECC depending on the bound ES. However, it is clear that editing of several ESs defined by one gRNA and pan-editing of mRNAs requiring multiple gRNAs requires a dynamic machinery involving more than RECC alone.
Multi-core processing: the MRB1 and other complexes
While the core editing activities encapsulated by RECC can be observed in vitro, the whole process of decrypting the ORFs of the pan-edited mRNAs that require a cascade of gRNAs cannot. Furthermore, RECC in vitro editing requires an already annealed dsRNA substrate, indicating that other factors are needed for the recruitment of one or both types of substrate RNAs to the complex for processing and/or other activities. The identification of molecules mediating such roles was started with the discovery of a dynamic collection of ~31 proteins with an association with RNA editing. These proteins were initially called either the mitochondrial RNA binding complex 1 (MRB1) (Hashimi et al., Reference Hashimi, Ziková, Panigrahi, Stuart and Lukeš2008; Panigrahi et al., Reference Panigrahi, Ziková, Dalley, Acestor, Ogata, Anupama, Myler and Stuart2008) or guide RNA binding complex (GRBC) (Weng et al., Reference Weng, Aphasizheva, Etheridge, Huang, Wang, Falick and Aphasizhev2008), the latter designation later ascribed to a sub-complex (see below) and replaced with RNA editing substrate binding complex (RESC) (Aphasizheva et al., Reference Aphasizheva, Zhang, Wang, Kaake, Huang, Monti and Aphasizhev2014). This complex will be referred to herein as MRB1 (Ammerman et al., Reference Ammerman, Downey, Hashimi, Fisk, Tomasello, Faktorová, Kafková, King, Lukeš and Read2012).
Further refinement of MRB1 architecture has revealed that it is made up of two sub-complexes with different roles in RNA editing (Ammerman et al., Reference Ammerman, Downey, Hashimi, Fisk, Tomasello, Faktorová, Kafková, King, Lukeš and Read2012; Aphasizheva et al., Reference Aphasizheva, Zhang, Wang, Kaake, Huang, Monti and Aphasizhev2014). Persistent in all reported MRB1 purifications are seven proteins that make up the MRB1 core (Read et al., Reference Read, Lukeš and Hashimi2016). The paralogous gRNA associated proteins (GAPs) 1 and 2 form a heterotetramer that binds gRNAs, a requisite for their stability, are the only verified RNA-binding proteins of the MRB1 core (Weng et al., Reference Weng, Aphasizheva, Etheridge, Huang, Wang, Falick and Aphasizhev2008; Hashimi et al., Reference Hashimi, Čičová, Novotná, Wen and Lukeš2009). Thus, the editing of cis-gRNA-containing COX2 is not affected by their RNAi-silencing (Hashimi et al., Reference Hashimi, Čičová, Novotná, Wen and Lukeš2009). Knockdown of the other core proteins does not destabilize gRNAs but appears to affect RNA editing initiation (Acestor et al., Reference Acestor, Panigrahi, Carnes, Zíková and Stuart2009; Ammerman et al., Reference Ammerman, Tomasello, Faktorová, Kafková, Hashimi, Lukeš and Read2013; Huang et al., Reference Huang, Faktorová, Křížová, Kafková, Read, Lukeš and Hashimi2015). Thus, it has been proposed that the MRB1 core plays a role in editing initiation (Read et al., Reference Read, Lukeš and Hashimi2016), although a general effect of MRB1 core ablation on gRNA utilization could be masked by an impaired gRNA phenotype. Since the GAP1/2 heterotetramer seems to have an extra-MRB1 localization, it may be involved in gRNA delivery to the editing reaction center, where these molecules pair with their cognate mRNAs.
The RNA editing mediator complex makes up the other major sub-complex of MRB1. It contains several RNA binding proteins, such as TbRGG2 (Ammerman et al., Reference Ammerman, Presnyak, Fisk, Foda and Read2010; Foda et al., Reference Foda, Downey, Fisk and Read2012), as well novel RNA binding proteins such as MRB8180 (Simpson et al., Reference Simpson, Bruno, Chen, Lott, Tylec, Bard, Sun, Buck and Read2017) or paralogues MRB8170 and MRB4160 (Kafková et al., Reference Kafková, Ammerman, Faktorová, Fisk, Zimmer, Sobotka, Read, Lukeš and Hashimi2012; Dixit et al., Reference Dixit, Muller-McNicoll, David, Zarnack, Ule, Hashimi and Lukeš2017). Ablation of these subunits preferentially leads to a stalling of pan-editing, which requires a cascade of gRNAs for its 3′–5′ progression (Fisk et al., Reference Fisk, Ammerman, Presnyak and Read2008; Kafková et al., Reference Kafková, Ammerman, Faktorová, Fisk, Zimmer, Sobotka, Read, Lukeš and Hashimi2012), suggesting a role of this sub-complex in mediating this process.
It has been proposed that the two sub-complexes that make up MRB1 together with the core catalytic RECC represent the true editosome holoenzyme (Aphasizheva et al., Reference Aphasizheva, Zhang, Wang, Kaake, Huang, Monti and Aphasizhev2014; Aphasizheva and Aphasizhev, Reference Aphasizheva and Aphasizhev2016). Certainly, the demonstrated roles of each of these modules can together account for the predicted machinery needed to decrypt a pan-edited mRNA. The reader can explore recent reviews dedicated to trypanosome RNA editing for more detailed discussions about the proteins and complexes involved in this fascinating phenomenon (Aphasizhev and Aphasizheva, Reference Aphasizhev and Aphasizheva2011; Hashimi et al., Reference Hashimi, Zimmer, Ammerman, Read and Lukes2013; Read et al., Reference Read, Lukeš and Hashimi2016).
The abundant mitochondrial RNA-binding proteins (MRP) 1 and 2 form a hetero-tetrameric complex with an electropositive surface that facilitates binding of the negatively charged sugar–phosphate backbone of RNA (Schumacher et al., Reference Schumacher, Karamooz, Zíková, Trantirek and Lukeš2006). Upon binding to the MRP1/2 complex, the RNA bases extrude out to be available for annealing complementary RNA, which is consistent with a proposed role in gRNA:mRNA annealing (Müller et al., Reference Müller, Lambert and Göringer2001; Zíková et al., Reference Zíková, Kopečná, Schumacher, Stuart, Trantírek and Lukeš2008a). A Nudix hydrolase was also pulled down with MRP1 that was later found to be part of the multiprotein mitochondrial edited RNA stability factor 1, MERS1 (Weng et al., Reference Weng, Aphasizheva, Etheridge, Huang, Wang, Falick and Aphasizhev2008). A recently discovered complex containing the terminal uridylyl-transferase (Aphasizhev et al., Reference Aphasizhev, Aphasizheva and Simpson2003) and a homologue of a yeast 3′–5′ exonuclease (Mattiacio and Read, Reference Mattiacio and Read2008) appears necessary for gRNA maturation (Suematsu et al., Reference Suematsu, Zhang, Aphasizheva, Monti, Huang, Wang, Costello and Aphasizhev2016).
Mitochondrial protein synthesis and the mRNA recognition problem
While the large body of evidence indicated that mitochondrial protein synthesis is responsible for the production of indispensable subunits of the respiratory complexes, its biochemical purification was problematic because kinetoplast-encoded polypeptides are extremely hydrophobic (Speijer et al., Reference Speijer, Breek, Muijsers, Hartog, Berden, Albracht, Samyn, Van Beeumen and Benne1997). As of today, at least four proteins were confirmed as products of mitochondrial translation (Horváth et al., Reference Horváth, Nebohacova, Lukeš and Maslov2002; Škodová-Sveráková et al., Reference Škodová-Sveráková, Horváth and Maslov2015).
Pre-edited and partially edited transcripts are relatively abundant in the steady-state RNA population. Since they cannot be productively translated, it is likely that there is a mechanism allowing for an exclusive recognition of fully edited, translation-competent mRNAs. It was recognized early that this mechanism cannot be reduced to a simple creation of the initiation codon or a Shine-Dalgarno-like sequence. Yet, a reasonable possibility was that editing creates some form of a translatability hallmark on the mRNA's 5′-end, in particular, because the arrival of editing at the 5′-end indicates that the entire sequence downstream has been edited and is, therefore, translatable. Although the mechanisms involved have not yet been fully elucidated, there has been a significant progress in this direction over the last several years.
Early investigations showed that fully edited mRNAs possess two types of 3′-end poly(A)-tails: the short, ~20–30 nt and the long, ~200–300 nt, while pre-edited and partially edited mRNAs contain only a short tail (Bhat et al., Reference Bhat, Koslowsky, Feagin, Smiley and Stuart1990; Kao and Read, Reference Kao and Read2005). Subsequently, it was shown that conditional upon completion of editing, the initial short tail is extended to become a long A/U heteropolymer (Etheridge et al., Reference Etheridge, Aphasizheva, Gershon and Aphasizhev2008). This reaction is performed by a protein complex composed of two catalytic proteins (KPAP1, a poly(A) polymerase, and RET1, an uridylyl transferase) and two auxiliary proteins (KPAF1 and KPAF2) (Aphasizheva et al., Reference Aphasizheva, Maslov, Wang, Huang and Aphasizhev2011). The latter belong to the large family of PPR (pentatricopeptide repeat) proteins, which are relatively abundant in trypanosomatids. Discovered in plants, these proteins are involved in numerous aspects of mRNA maturation and translation in plants organelles, and they proved to play very important roles in the kinetoplasts as well (Aphasizhev and Aphasizheva, Reference Aphasizhev and Aphasizheva2013). Inactivation of KPAF1 by RNAi resulted in a loss of the long (A/U)-tails, disruption of the mRNA interaction with the mitoribosomes and the cessation of the COI and Cyb synthesis. An attractive hypothesis that some of these PPR proteins may act as mRNA-specific translational activators was supported by a differential effect on the (A/U)-tailing and translation of COI and Cyb polypeptides caused by inactivation of KRIPP1 and KRIPP8 ribosomal PPR proteins in T. brucei (Aphasizheva et al., Reference Aphasizheva, Maslov, Qian, Huang, Wang, Costello and Aphasizhev2016). These proteins are components of a 45S complex, which also contains the 9S SSU rRNA, a set of small subunit ribosomal proteins and several additional PPR proteins (Maslov et al., Reference Maslov, Spremulli, Sharma, Bhargava, Grasso, Falick, Agrawal, Parker and Simpson2007). This complex, termed 45S SSU*, is abundant in procyclic T. brucei, but downregulated in its bloodstream stage (Ridlon et al., Reference Ridlon, Škodová, Pan, Lukeš and Maslov2013). Its disruption in procyclics abolished the poly(A/U)-tailing and translation of several mRNAs, which are expressed during this stage of the life cycle, but did not affect constitutively expressed products such as RPS12 and A6 (Ridlon et al., Reference Ridlon, Škodová, Pan, Lukeš and Maslov2013; Aphasizheva et al., Reference Aphasizheva, Maslov, Qian, Huang, Wang, Costello and Aphasizhev2016), suggesting that 45S SSU* complex is involved in the developmental regulation of mitochondrial translation in this species. Although details of this process remain unknown, the available data allow to speculate that a specific cis-signal is created upon completion of editing and the mRNA's 5′-end is recognized by an mRNA-specific PPR protein. This protein, acting as an mRNA specific translation activator, in turn mediates poly(A/U) tailing and recognition of the translation competent mRNA by the 45S SSU* complex. The steady-state level of kinetoplast-mitochondrial 50S ribosomes is low in comparison to ribosomal large subunits (Maslov et al., Reference Maslov, Sharma, Butler, Falick, Gingery, Agrawal, Spremulli and Simpson2006; Ridlon et al., Reference Ridlon, Škodová, Pan, Lukeš and Maslov2013), leading to a hypothesis that the active translation complex, which sediments at >80S, each time assembles de novo by association of the 40S large ribosomal subunit with the mRNA recognition complex.
The structure of the 50S Leishmania mitochondrial ribosomes has been investigated in detail by cryoelectron microscopy (Sharma et al., Reference Sharma, Booth, Simpson, Maslov and Agrawal2009). Surprisingly, the overall morphology of the 50S monosome appears remarkably eubacterial in spite of the drastic differences in the RNA and protein structure and composition. Indeed, the sizes of the 9S and 12S rRNAs are substantially smaller and their secondary structure lacks several stem-loop elements present in their eubacterial counterparts (Eperon et al., Reference Eperon, Janssen, Hoeijmakers and Borst1983; de la Cruz et al., Reference de la Cruz, Lake, Simpson and Simpson1985a, Reference de la Cruz, Simpson, Lake and Simpson1985b). The protein content represents a mixture of the conserved ribosomal and trypanosomatid-specific proteins (Maslov et al., Reference Maslov, Sharma, Butler, Falick, Gingery, Agrawal, Spremulli and Simpson2006; Reference Maslov, Spremulli, Sharma, Bhargava, Grasso, Falick, Agrawal, Parker and Simpson2007; Zíková et al., Reference Zíková, Panigrahi, Dalley, Acestor, Anupama, Ogata, Myler and Stuart2008b; Aphasizheva et al., Reference Aphasizheva, Maslov, Wang, Huang and Aphasizhev2011). In cryoelectron microscopy model, the missing RNA masses are only partially replaced by proteins resulting in an overall porous structure of the mitoribosome. A number of the functionally important regions, such as the mRNA and tRNA paths, and nascent polypeptide exit channel contain trypanosomatid-specific proteins or show other peculiarities, apparently reflecting the idiosyncratic modus operandi of these ribosomes, including its resistance to most inhibitors of protein synthesis (Sharma et al., Reference Sharma, Booth, Simpson, Maslov and Agrawal2009; Hashimi et al., Reference Hashimi, Kaltenbrunner, Zíková and Lukeš2016).
Mitoproteome
Mostly because of RNA editing and kDNA, the T. brucei mitochondrion belongs to the best studied organelles of unicellular eukaryotes. As a consequence, a high-quality mitoproteome became available (Panigrahi et al., Reference Panigrahi, Ogata, Zíková, Anupama, Dalley, Acestor, Myler and Stuart2009) and was used for identification of novel protein functions. The most prominent case is finding a protein responsible for the import of Ca2+ into the mitochondrion, an activity known for decades. Yet, the protein responsible for Ca2+ uptake, called the mitochondrial calcium uniporter (MCU), remained elusive until recently. It was the comparison of numerous mitochondrial profiles of organisms, known to either possess or lack this capacity, which facilitated discovery of the MCU (Baughman et al., Reference Baughman, Perocchi, Girgis, Plovanich, Belcher-Timme, Sancak, Bao, Strittmatter, Goldberger, Bogorad, Koteliansky and Mootha2011; Docampo and Lukeš, Reference Docampo and Lukeš2012).
Interestingly, this was not the only case. The prominent absence of several conserved proteins in the T. brucei mitoproteome was used in phylogenetic profiling, which resulted in identification of several novel assembly factors of the human respiratory complex I (Pagliarini et al., Reference Pagliarini, Calvo, Chang, Sheth, Vafai, Ong, Walford, Sugiana, Boneh, Chen, Hill, Vidal, Evans, Thorburn, Carr and Mootha2008). It is safe to predict that by virtue of being the only mitochondrion in the cell and by its significant functional and structural up- and downregulation throughout the life cycle (Zíková et al., Reference Zíková, Verner, Nenarokova, Michels and Lukeš2017), kinetoplast is particularly suitable for studies of processes that control mitochondrial functions and will provide important insight in this respect.
Organelles
Glycosomes
Virtually all eukaryotic cells have peroxisomes, i.e. microbodies involved in catabolism of long chain fatty acids, branched chain fatty acids, D-amino acids, polyamines, reduction of reactive oxygen species (ROS), specifically hydrogen peroxide, and biosynthesis of plasmalogen ether phospholipids. In trypanosomatids, glycolysis is associated with specialized peroxisomes called glycosomes, containing six enzymes involved in the early part of the glycolytic pathway, and two enzymes of glycerol metabolism (Opperdoes and Borst, Reference Opperdoes and Borst1977). In addition, trypanosomatid glycosomes are involved in gluconeogenesis, NADPH production via the glucose-6-phosphate dehydrogenase enzymes (Heise and Opperdoes, Reference Heise and Opperdoes1999), purine salvage and phosphate metabolism (Szöör et al., Reference Szöör, Haanstra, Gualdrón-López and Michels2014; Gabaldón et al., Reference Gabaldón, Ginger and Michels2016). None of the human-infective trypanosomatids (i.e. Leishmania spp., T. brucei or T. cruzi) possess a gene for the typical peroxisomal marker enzyme, catalase (Kraeva et al., Reference Kraeva, Horáková, Kostygov, Kořený, Butenko, Yurchenko and Lukeš2017). Only monoxenous Crithidia and Leptomonas spp. have a catalase gene (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016), although the enzyme is not present in peroxisomes, but in the cytosol (Souto-Padron and de Souza, Reference Souto-Padron and de Souza1982). Interestingly, the related cryptobiids have peroxisomes/glycosomes with catalase activity (Opperdoes et al., Reference Opperdoes, Nohýnková, Van Schaftingen, Lambeir, Veenhuis and Van Roy1988; Ardelli et al., Reference Ardelli, Witt and Woo2000), while B. saltans, the closest known bodonid relative of trypanosomatids, lacks this gene.
The presence of an NADP-dependent isocitrate dehydrogenase and one of the four Fe-superoxide dismutase isoenzymes in glycosomes suggest that sufficient ROS protection mechanisms must be present in these organelles (Dufernez et al., Reference Dufernez, Yernaux, Gerbod, Noël, Chauvenet, Wintjens, Edgcomb, Capron, Opperdoes and Viscogliosi2006). However, enzymes of the glyoxylate cycle, reported to be present in the peroxisomes of ciliates (Simon et al., Reference Simon, Martin and Mukkada1978) and two other typical peroxisomal marker enzymes, D-amino acid oxidase and 2-hydroxy acid oxidase, were not detected in any trypanosomatids, or B. saltans genomes (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016).
Many orthologues of glycosomal proteins well-characterized in trypanosomatids were recently identified in B. saltans (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016) suggesting their peroxisomes fulfill functions similar to those of trypanosomatid glycosomes. For a detailed account of the functions of glycosomes in trypanosomatid the reader is referred to recent papers (Opperdoes, Reference Opperdoes1987; Opperdoes and Szikora, Reference Opperdoes and Szikora2006; Vertommen et al., Reference Vertommen, Van Roy, Szikora, Rider, Michels and Opperdoes2008; Haanstra et al., Reference Haanstra, González-Marcano, Gualdrón-López and Michels2016).
Traffic of solutes between cytosol and glycosomes
Solutes, such as small metabolites, cofactors, and acyl-CoAs, all seem to be translocated by specific transporter molecules, such as ATP-binding cassette (ABC) transporters and membrane channels. Three ABC transporters, called Glycosomal ABC transporters 1–3 (GAT1-3), have been identified in the glycosomal membrane of T. brucei, where they mediate ATP-dependent uptake of solutes from the cytosol into the glycosomal matrix. GAT1 was shown to transport primarily oleoyl-CoA (Igoillo-Esteve et al., Reference Igoillo-Esteve, Mazet, Deumer, Wallemacq and Michels2011). Smaller solutes, such as glycolytic intermediates, probably cross the membrane through several types of pore-forming channels (Gualdron-López et al., Reference Gualdron-López, Vapola, Miinalainen, Hiltunen, Michels and Antonenkov2012).
The glycosome as an example of mathematical modelling
The long history of quantitative research and the detailed knowledge about the enzymes of carbohydrate metabolism, the reactions they catalyse, and their compartmentation within the glycosomes, has allowed one to construct a reliable kinetic computer model of trypanosome glycolysis (Bakker et al., Reference Bakker, Michels, Opperdoes and Westerhoff1997; Reference Bakker, Westerhoff, Opperdoes and Michels2000; Haanstra et al., Reference Haanstra, van Tuijl, Kessler, Reijnders, Michels, Westerhoff, Parsons and Bakker2008). Owing to this kinetic model, African trypanosomes have emerged as promising unicellular model organisms for the next generation of systems biology. The results are compiled in ‘Silicon Trypanosome’, a comprehensive, experiment-based, multi-scale mathematical model of trypanosome physiology (Bakker et al., Reference Bakker, Krauth-Siegel, Clayton, Matthews, Girolami, Westerhoff, Michels, Breitling and Barrett2010). It is anticipated that quantitative modelling enabled by the ‘Silicon Trypanosome’ will play a key role in selecting the most suitable targets for developing new anti-parasite drugs.
Acidocalcisomes
Acidocalcisomes were first discovered in trypanosomes (Docampo et al., Reference Docampo, de Souza, Miranda, Rohloff and Moreno2005). They are 100 to and 200 nm in diameter electron-dense acidic organelles serving as the primary calcium (Ca2+) reservoir, that is also rich in phosphate in the form of orthophosphate (Pi), pyrophosphate (PPi) and polyphosphate (polyP) (Lander et al., Reference Lander, Cordeiro, Huang and Docampo2016). Their internal acidity is maintained by proton pumps such as the vacuolar proton pyrophosphatase (V-H + -PPase, or VP1), the vacuolar proton ATPase (V-H + -ATPase), or both (Docampo, Reference Docampo2016). In addition to a number of protein pumps and antiporters, including aquaporins, the acidocalcisomal membranes contain various ATPases and Ca2+/H+ and Na+/H+ antiporters, suggesting a complex energetic requirement for their maintenance. The acidocalcisomes also play a role in autophagy and osmoregulation (Docampo, Reference Docampo2016; Docampo and Huang, Reference Docampo and Huang2016). When T. cruzi is exposed to an osmotic shock, these organelles located in the vicinity of the contractile vacuole fuse with it, thereby increasing its osmolarity. As a consequence, water from the cytoplasm enters the vacuole for expulsion (Rohloff et al., Reference Rohloff, Montalvetti and Docampo2004). The release of an important second messenger Ca2+ from intracellular stores is controlled by the inositol 1,4,5-trisphosphate receptor located inside the acidocalcisomes, while a plasma membrane Ca2+-ATPase controls the cytosolic Ca2+ level. In trypanosomatids with an intracellular life stage, Ca2+signalling is proposed to govern host cell invasion (Docampo and Huang, Reference Docampo and Huang2016).
Highly flexible flagellum
All trypanosomatids are equipped with a single flagellum (although, there is an ‘amastigote’ stage in some life cycles, characterized by an extremely short flagellum), which represents the most prominent morphological difference from their bodonid kins with two flagella (Adl et al., Reference Adl, Simpson, Lane, Lukeš, Bass, Bowser, Brown, Burki, Dunthorn, Hampl, Heiss, Hoppenrath, Lara, Le Gall, Lynn, McManus, Mitchell, Mozley-Stanridge, Parfrey, Pawlowski, Rueckert, Shadwick, Schoch, Smirnov and Spiegel2012). The flagellum length is highly variable between and even within species, yet its structure is highly conserved and unique for this group of protists. It is also a highly flexible structure mostly involved in attachment, locomotion and environment sensing (Broadhead et al., Reference Broadhead, Dawe, Farr, Griffiths, Hart, Portman, Shaw, Ginger, Gaskell, McKean and Gull2006; Hughes et al., Reference Hughes, Ralston, Hill and Zhou2012). During the life cycle, the flagellum is subject to substantial restructuring to adapt to different functions (Ginger et al., Reference Ginger, Portman and McKean2008). In the best-studied species, T. brucei, the flagellar motility is required for cell division, transmission via a vector, immune evasion (Engstler et al., Reference Engstler, Pfohl, Herminghaus, Boshart, Wiegertjes, Heddergott and Overath2007) and is also intimately associated with the vital flagellar pocket structure (Field and Carrington, Reference Field and Carrington2009). Recently, additional functions of this dexterous cellular component such as the production of the extracellular vesicles, which may mediate interaction with the vertebrate host, have been described (Szempruch et al., Reference Szempruch, Sykes, Kieft, Dennison, Becker, Gartrell, Martin, Nakayasu, Almeida, Hajduk and Harrington2016). Furthermore, protein exchange between two trypanosomes seems to occur by flagellar membrane exchange, and both short and long-term fusions have been observed in cultured trypanosomes (Imhof et al., Reference Imhof, Fragoso, Hemphill, von Schubert, Li, Legant, Betzig and Roditi2016).
The trypanosomatid flagellum, responsible for motility, contains the classical 9 + 2 axoneme (Ginger et al., Reference Ginger, Portman and McKean2008). The 9 + 0 axoneme has been observed in the amastigote stages of Leishmania spp., where the flagellum is likely to be more engaged in sensing and signalling (Wheeler et al., Reference Wheeler, Gluenz and Gull2015). A characteristic feature of the trypanosomatid flagellum is the paraflagellar rod, an extra-axonemal structure. It is very prominent in some species (Yurchenko et al., Reference Yurchenko, Lukeš, Jirků, Zeledon and Maslov2006a; Maslov et al., Reference Maslov, Yurchenko, Jirků and Lukeš2010) and almost invisible in others (Yurchenko et al., Reference Yurchenko, Votýpka, Tesařová, Klepetková, Kraeva, Jirků and Lukeš2014), with the arrangement of thin and thick filaments also being species-specific (Gadelha et al., Reference Gadelha, Wickstead, de Souza, Gull and Cunha-e-Silva2005; Sant'Anna et al., Reference Sant'Anna, Campanati, Gadelha, Lourenco, Labati-Terra, Bittencourt-Silvestre, Benchimol, Cunha-e-Silva and De Souza2005). So far, about 30 proteins have been identified as components of the T. brucei paraflagellar rod (Portman and Gull, Reference Portman and Gull2010). Their ablation or deletion often, but not always results in a dramatic decrease in flagellar beating frequency. Interestingly, while in the procyclic stage of T. brucei RNAi-mediated downregulation of paraflagellar proteins occasionally causes cytokinesis defects (Farr and Gull, Reference Farr and Gull2009), in the bloodstream stage flagellar motility seems to be invariably essential for viability (Broadhead et al., Reference Broadhead, Dawe, Farr, Griffiths, Hart, Portman, Shaw, Ginger, Gaskell, McKean and Gull2006), and hence is of medical relevance. It was proposed that the paraflagellar rod might be a site for integrating external signals detected by the flagellum (Portman and Gull, Reference Portman and Gull2010).
During their life cycle, the cell shape of most trypanosomatids undergoes dramatic morphological changes. These are controlled by a specialized cytoskeletal structure termed the flagellum attachment zone. It laterally attaches the flagellum to the cytoskeleton and seems to play a key role in determining trypanosomatid morphology (Sunter et al., Reference Sunter, Varga, Dean and Gull2015). The flagellum attachment zone ranges from an extended form in trypomastigotes to a very short one in promastigotes (Wheeler et al., Reference Wheeler, Gluenz and Gull2015). Recently, the early-branching Paratrypanosoma confusum was shown to restructure its flagellum during the life cycle from a promastigote with a long flagellum to an amastigote-like stage with no external flagellum, and then to a cell in which the flagellum is remodeled into a thin attachment pad (Skalický et al., Reference Skalický, Dobáková, Wheeler, Tesařová, Flegontov, Jirsová, Votýpka, Yurchenko, Ayala and Lukeš2017). Hence, the enormous flexibility of the flagellum and related structures seems to be an ancestral feature that might have predetermined trypanosomatids for their evolutionary expansion.
Gene exchange
Cellular mechanisms
The importance of the question of whether the binary fission is the only (or at least the predominant) reproduction mode in trypanosomatids goes far beyond being purely academic. The existence of meiosis and potential for gamete fusion or a similar type of sexual process would determine if trypanosomes are capable of gene exchange as opposed to strictly asexual (clonal) propagation in natural populations (Tait, Reference Tait1980). This question is central to our understanding of the origin and spread of pathogenic traits with obvious implications for epidemiology and treatment.
A sexual process was first demonstrated in African trypanosomes in hybridization experiments during co-infection of tsetse flies with two parental clonal lines of T. brucei (Jenni et al., Reference Jenni, Marti, Schweizer, Betschart, Le Page, Wells, Tait, Paindavoine, Pays and Steinert1986). Selection of the hybrids for double drug resistance had greatly facilitated identification of the recombinant progeny as the mating, which occurs in salivary glands of infected tsetse flies, was found to be non-obligatory (Gibson and Whittington, Reference Gibson and Whittington1993; Gibson and Bailey, Reference Gibson and Bailey1994). Interestingly, while kDNA minicircles were inherited from both parents, the maxicircles initially appeared to be inherited uniparentally (Gibson and Garside, Reference Gibson and Garside1990). However, subsequently it was demonstrated that the maxicircle inheritance is biparental, but the initial heteroplasmic state is rapidly eliminated due to a stochastic segregation of maxicircles during mitotic divisions (Turner et al., Reference Turner, Hide, Buchanan and Tait1995). The inheritance pattern of nuclear chromosomes was biparental and consistent with Mendelian segregation and independent assortment, providing further proof for the meiosis involvement (Turner et al., Reference Turner, Sternberg, Buchanan, Smith, Hide and Tait1990; Gibson and Garside, Reference Gibson and Garside1991; MacLeod et al., Reference MacLeod, Tweedie, McLellan, Hope, Taylor, Cooper, Sweeney, Turner and Tait2005).
Further insights into details of the sexual process were obtained upon the development of green and red fluorescent parental lines, allowing the detection of individual hybrid trypanosomes by yellow fluorescence directly in the salivary glands of double-infected tsetse flies (Gibson et al., Reference Gibson, Peacock, Ferris, Williams and Bailey2008). Being epimastigotes, the hybrids were observed exclusively in the salivary glands as soon as the parental cells have reached this compartment. Moreover, by using fluorescent tagging, the expression of three meiosis-specific genes was found to take place during a certain time window in all tested T. brucei subspecies, indicating that all are capable of gene exchange (Peacock et al., Reference Peacock, Ferris, Sharma, Sunter, Bailey, Carrington and Gibson2011, Reference Peacock, Ferris, Bailey and Gibson2014b). With the aim to identify products of the meiotic cell division (gametes), the green and red fluorescent cells were recovered from the salivary glands of infected flies at the peak of meiosis-specific gene expression and mixed ex vivo for microscopic examination (Peacock et al., Reference Peacock, Bailey, Carrington and Gibson2014a). Putative gametes were observed as haploid fluorescent red and green promastigote-like cells with a single or two kinetoplasts and a single long flagellum. These cells would interact by intertwining their flagella and apparently undergoing fusion as indicated by the appearance of yellow fluorescent cells shortly thereafter.
The question about the existence of mating types in trypanosomes remains open. They are able to undergo intraclonal mating (self-fertilization), although it is far less efficient compared to mating between different parental cells, indicating either the absence of mating types or a rather unconventional mating type system (Turner et al., Reference Turner, Sternberg, Buchanan, Smith, Hide and Tait1990; Tait et al., Reference Tait, Buchanan, Hide and Turner1996; Peacock et al., Reference Peacock, Ferris, Bailey and Gibson2009). Recent analyses confirmed that T. brucei crosses are inconsistent with a ‘two mating types’ system (Peacock et al., Reference Peacock, Ferris, Bailey and Gibson2014b). It was further hypothesized that these mating types may be controlled by multiple alleles of variable efficiency and there exists a potential for mating type switching during development in tsetse flies (Peacock et al., Reference Peacock, Bailey, Carrington and Gibson2014a). These studies have established that trypanosomes have an intrinsic ability to undergo meiosis and to produce hybrids by gametic fusion, albeit actual gene exchange is not mandatory in the T. brucei life cycle (Gibson, Reference Gibson2015). Both selfing and interclonal mating are possible, and the sexual process is not limited to a particular subgroup of African trypanosomes but represents a general property of these parasites.
Less is known about meiosis and gene exchange in other trypanosomatids. Meiosis-specific genes are also present in the genomes of Leishmania species and, perhaps, most other trypanosomatids (Ramesh et al., Reference Ramesh, Malik and Logsdon2005; Speijer et al., Reference Speijer, Lukeš and Eliáš2015), as they were recently identified in the genomes of two Leptomonas spp. (Kraeva et al., Reference Kraeva, Butenko, Hlaváčová, Kostygov, Myškova, Grybchuk, Leštinová, Votýpka, Volf, Opperdoes, Flegontov, Lukeš and Yurchenko2015; Flegontov et al., Reference Flegontov, Butenko, Firsov, Kraeva, Eliáš, Field, Filatov, Flegontova, Gerasimov, Hlaváčová, Ishemgulova, Jackson, Kelly, Kostygov, Logacheva, Maslov, Opperdoes, O'Reilly, Sádlová, Ševčíková, Venkatesh, Vlček, Volf, Votýpka, Záhonová, Yurchenko and Lukeš2016). Experimental evidence for hybrid formation in the sand fly vector has originally been obtained for Leishmania major (Akopyants et al., Reference Akopyants, Kimblin, Secundino, Patrick, Peters, Lawyer, Dobson, Beverley and Sacks2009). Most biparental hybrid clones, selected by double drug resistance, were diploid, but some were triploid, and the inheritance of the kDNA maxicircles appeared to be uniparental. The frequency of hybridization was rather low, at the level of ~10−5. Subsequently, using a double (red-green) fluorescence system in L. donovani (Sádlová et al., Reference Sádlová, Yeo, Seblová, Lewis, Mauricio, Volf and Miles2011) it was shown that the hybrid cells appear as procyclic promastigotes (but see below) in the midgut of infected sand flies as early as day 2 post-blood meal. The hybrid cell lines could not be recovered precluding their further characterization. More recently, numerous interclonal hybrids were obtained for L. major (Inbar et al., Reference Inbar, Akopyants, Charmoy, Romano, Lawyer, Elnaiem, Kauffmann, Barhoumi, Grigg, Owens, Fay, Dobson, Shaik, Beverley and Sacks2013) and two intraclonal hybrids were obtained for L. infantum (Calvo-Alvarez et al., Reference Calvo-Alvarez, Alvarez-Velilla, Jimenez, Molina, Perez-Pertejo, Balana-Fouce and Reguera2014). While the L. major hybrids were mostly diploid with the frequent occurrence of triploid and even some tetraploid cell lines, the two L. infantum hybrid clones were triploid. Interestingly, these were able to infect mice. While diploid hybrids are consistent with the model involving meiosis and a haploid gametic fusion, the triploid cells would be produced by a fusion of a haploid and a diploid cell, as was suggested for triploid hybrids formed in some crosses of T. brucei (Gibson et al., Reference Gibson, Garside and Bailey1992). The timing of hybrid formation in L. major suggested that nectomonads, rather than procyclic promastigotes, represent the mating-competent developmental stage (Inbar et al., Reference Inbar, Akopyants, Charmoy, Romano, Lawyer, Elnaiem, Kauffmann, Barhoumi, Grigg, Owens, Fay, Dobson, Shaik, Beverley and Sacks2013). Hybrid formation frequency suggested a lack of a strict mating type system in Leishmania. Overall, although many details of the sexual process in Leishmania still need to be elucidated and some of its aspects are likely to differ from their counterparts in T. brucei, it is clear that in both cases there is a solid evidence for sex based on meiosis and subsequent fusion of haploid gametes, which occurs in the insect vector.
Considering insect trypanosomatids, in Crithidia bombi, a parasite of bumblebees, there is evidence for a meiosis-related process with allele segregation and recombination, although the cellular mechanisms involved remain uncharacterized (Schmid-Hempel et al., Reference Schmid-Hempel, Salathe, Tognazzo and Schmid-Hempel2011; Cisarovsky and Schmid-Hempel, Reference Cisarovsky and Schmid-Hempel2014). The recently sequenced genome of this species (Schmid-Hempel et al., Reference Schmid-Hempel, Aebi, Barribeau, Kitajima, du Plessis, Schmid-Hempel and Zoller2018) will help in this regard.
A similar sexual process may also exist in T. cruzi as suggested by the presence of the conserved meiosis-specific genes (Ramesh et al., Reference Ramesh, Malik and Logsdon2005), although the limited experimental evidence obtained so far supports a different scenario (Gaunt et al., Reference Gaunt, Yeo, Frame, Stothard, Carrasco, Taylor, Mena, Veazey, Miles, Acosta, de Arias and Miles2003). Hybrid T. cruzi were formed exclusively during coinfection of a mammalian cell culture, representing the vertebrate stage of the life cycle, and not during passage through a triatomine bug vector. The hybrids were characterized by the inheritance of all parental alleles at most loci and massive aneuploidy. To explain these observations, a parasexual process has been implied, according to which nuclear fusion creates a tetraploid intermediate, that undergoes homologous recombination and partial genome reduction (Messenger and Miles, Reference Messenger and Miles2015). Still, the existence of a meiosis-related process and its role in the formation of the naturally occurring T. cruzi hybrid lineages remain an open question (Lewis et al., Reference Lewis, Llewellyn, Yeo, Acosta, Gaunt and Miles2011; Messenger and Miles, Reference Messenger and Miles2015).
Implications for population structure
A demonstration of a genetic recombination in laboratory settings, especially if the process is found to be non-obligatory, does not automatically entail its recognition as an important factor shaping the natural populations of that organism. It is difficult to overestimate the importance of the mode(s) of propagation of a parasite in nature [e.g. clonal, epidemic or panmictic (Smith et al., Reference Smith, Smith, O’Rourke and Spratt1993)] has for understanding its evolutionary trends, as well as the origin and spread of the disease it causes (Heitman, Reference Heitman2006). The main advantage of the strictly clonal mode is the possibility of a rapid propagation of the most successful gene combinations (or MLGs, multilocus genotypes), which are optimal (the fittest) under given conditions. However, the inevitable accumulation of deleterious mutations would lead to a decrease in fitness and, eventually, extinction – a situation known as Muller's ratchet. Introduction and spread of favourable mutations in populations can be achieved by a sexual process, although this comes at the cost of potentially disrupting the fittest MLGs by genetic recombination (Barton and Charlesworth, Reference Barton and Charlesworth1998). The population genetics of pathogenic trypanosomatids has, therefore, attracted significant attention (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2013; Reference Tibayrenc and Ayala2015; Reference Tibayrenc and Ayala2017; Ramirez and Llewellyn, Reference Ramirez and Llewellyn2014; Messenger and Miles, Reference Messenger and Miles2015; Rougeron et al., Reference Rougeron, De Meeus and Banuls2017).
Based on evidence against meiotic segregation of alleles (fixed heterozygocity, deviation from the Hardy–Weinberg expectation) and against genetic recombination (strong linkage disequilibrium, ubiquitous multilocus genotypes) observed in the natural populations of several parasitic protists, including trypanosomes and leishmanias, a ‘clonal theory’ was proposed (Tibayrenc et al., Reference Tibayrenc, Kjellberg and Ayala1990; Tibayrenc and Ayala, Reference Tibayrenc and Ayala1991). It postulates that in the absence of any consequential impact of gene exchange on a given population structure, ‘uniparental reproduction is, at least for the cases herein surveyed, predominant enough in natural populations to generate clones that are stable in space and time, even on an evolutionary time scale’ (Tibayrenc et al., Reference Tibayrenc, Kjellberg and Ayala1990). Stated this way, the clonal theory, while focusing on the importance of clonal reproduction for certain taxa or populations, does not necessarily exclude the occurrence of scenarios in which gene exchange, no matter how (in)frequent, would play a significant role. With time, the theory has evolved to become known as ‘predominantly clonal evolution’ (PCE), apparently to emphasize the long-term and large-scale implications of limited or absent genetic exchange.
In populations of T. cruzi, the showcase species for clonal theory, the predominantly clonal propagation mode was originally developed by analyses of isoenzyme electrophoretic patterns (MLEE) (Tibayrenc and Ayala, Reference Tibayrenc and Ayala1988), randomly amplified loci (Tibayrenc et al., Reference Tibayrenc, Neubauer, Barnabe, Guerrini, Skarecky and Ayala1993) and microsatellites (Oliveira et al., Reference Oliveira, Broude, Macedo, Cantor, Smith and Pena1998). These analyses revealed the existence of a complex population structure of these parasites (Miles et al., Reference Miles, Souza, Povoa, Shaw, Lainson and Toye1978; McDaniel and Dvorak, Reference McDaniel and Dvorak1993; Barnabe et al., Reference Barnabe, Brisse and Tibayrenc2000) with the existence of six major phylogenetic lineages (Brisse et al., Reference Brisse, Dujardin and Tibayrenc2000; Reference Brisse, Verhoef and Tibayrenc2001). The scale of genetic separation among these lineages was comparable with that of African trypanosomes or Leishmania spp., yet in the absence of the formal taxonomic status, the major lineages of T. cruzi were termed Discrete Typing Units (Tibayrenc, Reference Tibayrenc1998). Reflecting the evidence for genetic exchange in natural populations (Machado and Ayala, Reference Machado and Ayala2001; Brisse et al., Reference Brisse, Henriksson, Barnabe, Douzery, Berkvens, Serrano, De Carvalho, Buck, Dujardin and Tibayrenc2003), the term ‘near-clones’/‘near-clades’ has been proposed for them recently (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2012; Reference Tibayrenc and Ayala2015). Indeed, as four of these near-clades have a hybrid origin (Sturm et al., Reference Sturm, Vargas, Westenberger, Zingales and Campbell2003; Westenberger et al., Reference Westenberger, Barnabe, Campbell and Sturm2005; Lewis et al., Reference Lewis, Llewellyn, Yeo, Acosta, Gaunt and Miles2011), the strict clonality model is untenable. The PCE model postulates that although recombination in T. cruzi was important on a large evolutionary scale, it was unable to ‘prevent evolutionary divergence of the near-clades’ (Tibayrenc and Ayala, Reference Tibayrenc and Ayala2015).
Consistent with meiosis-based gene exchange being an inherent part of the life cycle in T. brucei, this process has been found to play a large role in shaping its natural populations. As two of its formal subspecies (T. b. rhodesiense and T. b. gambiense) are the causative agents of Human African Trypanosomiasis, gene exchange among those and non-infective subspecies (T. b. brucei) is important for understanding the origin and dynamics of disease foci (Gibson and Stevens, Reference Gibson and Stevens1999; Hide and Tait, Reference Hide and Tait2009). As described above, the relative importance of gene exchange vs clonality was not the same among different constituents of this species (MacLeod et al., Reference MacLeod, Tait and Turner2001a). By analysis of highly polymorphic minisatellite loci, it was demonstrated that East and South African human-infective T. b. rhodesiense is diverse and some isolates of this subspecies are genetically closer to local non-infective strains (regarded as the T. b. brucei subspecies) rather than to other infective strains (MacLeod et al., Reference MacLeod, Tait and Turner2001a, Reference MacLeod, Welburn, Maudlin, Turner and Tait2001c). This indicates that T. b. rhodesiense is just a host range variant of T. b. brucei, the populations of which are neither panmictic nor strictly clonal, but show evidence of limited gene exchange (epidemic structure) (MacLeod et al., Reference MacLeod, Tweedie, Welburn, Maudlin, Turner and Tait2000; Reference MacLeod, Turner and Tait2001b). Considering that human infectivity is defined by the presence of a single gene (serum resistance associated gene (SRA)) (De Greef and Hamers, Reference De Greef and Hamers1994; Xong et al., Reference Xong, Vanhamme, Chamekh, Chimfwembe, Van Den Abbeele, Pays, Van Meirvenne, Hamers, De Baetselier and Pays1998), the data strongly suggested that new strains of T. b. rhodesiense arise by genetic recombination spreading the SRA gene among local populations of T. b. brucei (Gibson et al., Reference Gibson, Backhouse and Griffiths2002; Balmer et al., Reference Balmer, Beadell, Gibson and Caccone2011). Subsequently, the idea of T. b. rhodesiense evolving from diverse genetic backgrounds of T. b. brucei has been supported by population genomics (Sistrom et al., Reference Sistrom, Evans, Bjornson, Gibson, Balmer, Maser, Aksoy and Caccone2014) and microsatellite studies, with the latter demonstrating genetic exchange occurring between some T. b. rhodesiense strains (Duffy et al., Reference Duffy, MacLean, Sweeney, Cooper, Turner, Tait, Sternberg, Morrison and MacLeod2013; Echodu et al., Reference Echodu, Sistrom, Bateta, Murilla, Okedi, Aksoy, Enyioha, Enyaru, Opiyo, Gibson and Caccone2015) and supporting the clonality of some others (Kato et al., Reference Kato, Alibu, Nanteza, Mugasa and Matovu2016).
The second pathogenic subspecies, the West African T. b. gambiense, has a different set of adaptations for human infectivity (Uzureau et al., Reference Uzureau, Uzureau, Lecordier, Fontaine, Tebabi, Homble, Grelard, Zhendre, Nolan, Lins, Crowet, Pays, Felu, Poelvoorde, Vanhollebeke, Moestrup, Lyngso, Pedersen, Mottram, Dufourc, Perez-Morga and Pays2013) and was found to form groups 1 and 2 by MLEE (Gibson, Reference Gibson1986; Godfrey et al., Reference Godfrey, Baker, Rickman and Mehlitz1990). Microsatellite locus typing has shown that group 1 is distinct, shows clear signs of strict clonality and is composed of a set of clades that occupy distinct geographic locations (Koffi et al., Reference Koffi, Solano, Barnabe, de Meeus, Bucheton, Cuny and Jamonneau2007, Reference Koffi, De Meeus, Bucheton, Solano, Camara, Kaba, Cuny, Ayala and Jamonneau2009; Morrison et al., Reference Morrison, Tait, McCormack, Sweeney, Black, Truc, Likeufack, Turner and MacLeod2008). Clonal evolution in group 1 trypanosomes was recently corroborated by a population genomics study demonstrating the independent accumulation of mutations in individual members of each homologous pair of chromosomes due to a lack of recombination, known as the ‘Meselson’ effect (Weir et al., Reference Weir, Capewell, Foth, Clucas, Pountain, Steketee, Veitch, Koffi, De Meeus, Kabore, Camara, Cooper, Tait, Jamonneau, Bucheton, Berriman and MacLeod2016). On the contrary, T. b. gambiense group 2 was found to be indistinguishable from local T. b. brucei, demonstrating evidence for gene exchange within and between human infective and non-infective trypanosomes (Capewell et al., Reference Capewell, Cooper, Duffy, Tait, Turner, Gibson, Mehlitz and Macleod2013). The reason for such a drastic difference between T. b. gambiense groups 1 and 2 remains unclear, especially because both groups possess and express meiosis-specific genes (Peacock et al., Reference Peacock, Ferris, Bailey and Gibson2014b). However, future population genomics studies may shed some more light. Indeed, the analysis of two T. b. rhodesiense genomes showed that these East African strains share some alleles with T. b. gambiense group 1, suggesting a gene flow between these subspecies in the past (Goodhead et al., Reference Goodhead, Capewell, Bailey, Beament, Chance, Kay, Forrester, MacLeod, Taylor, Noyes and Hall2013). It remains unclear if this was mediated by the local populations of T. b. brucei or occurred directly between the two pathogenic subspecies. In any case, the emerging picture presents a highly dynamic system, in which successful propagation is achieved by a combination of clonality and gene exchange.
Clonality was also proposed initially as the predominant propagation mode for Leishmania (Tibayrenc et al., Reference Tibayrenc, Kjellberg and Ayala1990; Banuls et al., Reference Banuls, Hide and Tibayrenc1999). Subsequently, a more complex picture has emerged in which both clonality and gene exchange play significant roles, called a ‘mixed-mating reproductive strategy’ (Rougeron et al., Reference Rougeron, De Meeus and Banuls2017). While significant inbreeding and clonality signatures were found in populations of L. braziliensis and L. guyanensis (Rougeron et al., Reference Rougeron, De Meeus, Hide, Waleckx, Bermudez, Arevalo, Llanos-Cuentas, Dujardin, De Doncker, Le Ray, Ayala and Banuls2009, Reference Rougeron, Banuls, Carme, Simon, Couppie, Nacher, Hide and De Meeus2011a; Kuhls et al., Reference Kuhls, Cupolillo, Silva, Schweynoch, Boite, Mello, Mauricio, Miles, Wirth and Schonian2013), the preponderance of clonality was stronger in studied populations of L. donovani (Rougeron et al., Reference Rougeron, De Meeus, Hide, Le Falher, Bucheton, Dereure, El-Safi, Dessein and Banuls2011b). A recent population genomics study of L. donovani from epidemic foci in India showed evidence for drug resistance having spread among populations by genetic recombination, as well as for clonal propagation of the major genetic groups under study (Imamura et al., Reference Imamura, Downing, Van den Broeck, Sanders, Rijal, Sundar, Mannaert, Vanaerschot, Berg, De Muylder, Dumetz, Cuypers, Maes, Domagalska, Decuypere, Rai, Uranw, Bhattarai, Khanal, Prajapati, Sharma, Stark, Schonian, De Koning, Settimo, Vanhollebeke, Roy, Ostyn, Boelaert, Maes, Berriman, Dujardin and Cotton2016). Thus in a way similar to T. brucei, Leishmania spp. illustrate how a successful parasite is able to utilize the advantages provided by each of the available propagation modes.
Adaptation of metabolism to parasitic lifestyle by gain and loss of genes
All Kinetoplastea share a number of unique metabolic characteristics. Most prominent are: (i) glycosomes (Opperdoes, Reference Opperdoes1987); (ii) a set of Pyr genes of the pyrimidine biosynthetic pathway with typical prokaryotic features (Opperdoes and Michels, Reference Opperdoes and Michels2007); (iii) ATP-dependent phosphofructokinase (PFK), strongly resembling bacterial pyrophosphate (PPi)-dependent PFKs, along with a PPi-dependent pyruvate phosphodikinase (Michels et al., Reference Michels, Chevalier, Opperdoes, Rider and Rigden1997; Cosenza et al., Reference Cosenza, Bringaud, Baltz and Vellieux2002); (iv) multiple phosphoglycerate kinases (Barros-Alvarez et al., Reference Barros-Alvarez, Caceres, Michels, Concepcion and Quinones2014) and two glyceraldehyde-phosphate dehydrogenases (Michels et al., Reference Michels, Marchand, Kohl, Allert, Wierenga and Opperdoes1991); (v) pyruvate kinase, allosterically regulated by the metabolic activator fructose-2,6-bisphosphate (van Schaftingen et al., Reference van Schaftingen, Opperdoes and Hers1985); (vi) trypanothione, rather than glutathione, as the major thiol involved in protection against the oxidative stress (Fairlamb et al., Reference Fairlamb, Blackburn, Ulrich, Chait and Cerami1985); (vii) synthesis of fatty acids via a unique set of elongases (Lee et al., Reference Lee, Stephens and Englund2007a), and (viii) a mitochondrial pathway for the ‘anaerobic’ excretion of acetate with net synthesis of ATP (van Hellemond et al., Reference van Hellemond, Opperdoes and Tielens1998). Thus, the last common ancestor of B. saltans and the trypanosomatids, which must have lived around 600 million years ago (Parfrey et al., Reference Parfrey, Lahr, Knoll and Katz2011; Lukeš et al., Reference Lukeš, Skalický, Týč, Votýpka and Yurchenko2014), had already acquired many genes of either bacterial or algal origin responsible for the aforementioned traits (Hannaert et al., Reference Hannaert, Bringaud, Opperdoes and Michels2003; Opperdoes and Coombs, Reference Opperdoes and Coombs2007; Opperdoes and Michels, Reference Opperdoes and Michels2007).
Comparison of the genome sequence of B. saltans (Jackson et al., Reference Jackson, Otto, Aslett, Armstrong, Bringaud, Schlacht, Hartley, Sanders, Wastling, Dacks, Acosta-Serrano, Field, Ginger and Berriman2016; Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016) with those available for a large number of trypanosomatids (Berriman et al., Reference Berriman, Ghedin, Hertz-Fowler, Blandin, Renauld, Bartholomeu, Lennard, Caler, Hamlin, Haas, Bohme, Hannick, Aslett, Shallom, Marcello, Hou, Wickstead, Alsmark, Arrowsmith, Atkin, Barron, Bringaud, Brooks, Carrington, Cherevach, Chillingworth, Churcher, Clark, Corton, Cronin, Davies, Doggett, Djikeng, Feldblyum, Field, Fraser, Goodhead, Hance, Harper, Harris, Hauser, Hostetler, Ivens, Jagels, Johnson, Johnson, Jones, Kerhornou, Koo, Larke, Landfear, Larkin, Leech, Line, Lord, Macleod, Mooney, Moule, Martin, Morgan, Mungall, Norbertczak, Ormond, Pai, Peacock, Peterson, Quail, Rabbinowitsch, Rajandream, Reitter, Salzberg, Sanders, Schobel, Sharp, Simmonds, Simpson, Tallon, Turner, Tait, Tivey, Van Aken, Walker, Wanless, Wang, White, White, Whitehead, Woodward, Wortman, Adams, Embley, Gull, Ullu, Barry, Fairlamb, Opperdoes, Barrell, Donelson, Hall, Fraser, Melville and El-Sayed2005; Ivens et al., Reference Ivens, Peacock, Worthey, Murphy, Aggarwal, Berriman, Sisk, Rajandream, Adlem, Aert, Anupama, Apostolou, Attipoe, Bason, Bauser, Beck, Beverley, Bianchettin, Borzym, Bothe, Bruschi, Collins, Cadag, Ciarloni, Clayton, Coulson, Cronin, Cruz, Davies, De Gaudenzi, Dobson, Duesterhoeft, Fazelina, Fosker, Frasch, Fraser, Fuchs, Gabel, Goble, Goffeau, Harris, Hertz-Fowler, Hilbert, Horn, Huang, Klages, Knights, Kube, Larke, Litvin, Lord, Louie, Marra, Masuy, Matthews, Michaeli, Mottram, Muller-Auer, Munden, Nelson, Norbertczak, Oliver, O’Neil, Pentony, Pohl, Price, Purnelle, Quail, Rabbinowitsch, Reinhardt, Rieger, Rinta, Robben, Robertson, Ruiz, Rutter, Saunders, Schafer, Schein, Schwartz, Seeger, Seyler, Sharp, Shin, Sivam, Squares, Squares, Tosato, Vogt, Volckaert, Wambutt, Warren, Wedler, Woodward, Zhou, Zimmermann, Smith, Blackwell, Stuart, Barrell and Myler2005; El-Sayed et al., Reference El-Sayed, Myler, Bartholomeu, Nilsson, Aggarwal, Tran, Ghedin, Worthey, Delcher, Blandin, Westenberger, Caler, Cerqueira, Branche, Haas, Anupama, Arner, Aslund, Attipoe, Bontempi, Bringaud, Burton, Cadag, Campbell, Carrington, Crabtree, Darban, da Silveira, de Jong, Edwards, Englund, Fazelina, Feldblyum, Ferella, Frasch, Gull, Horn, Hou, Huang, Kindlund, Klingbeil, Kluge, Koo, Lacerda, Levin, Lorenzi, Louie, Machado, McCulloch, McKenna, Mizuno, Mottram, Nelson, Ochaya, Osoegawa, Pai, Parsons, Pentony, Pettersson, Pop, Ramirez, Rinta, Robertson, Salzberg, Sanchez, Seyler, Sharma, Shetty, Simpson, Sisk, Tammi, Tarleton, Teixeira, Van Aken, Vogt, Ward, Wickstead, Wortman, White, Fraser, Stuart and Andersson2005a, Reference El-Sayed, Myler, Blandin, Berriman, Crabtree, Aggarwal, Caler, Renauld, Worthey, Hertz-Fowler, Ghedin, Peacock, Bartholomeu, Haas, Tran, Wortman, Alsmark, Angiuoli, Anupama, Badger, Bringaud, Cadag, Carlton, Cerqueira, Creasy, Delcher, Djikeng, Embley, Hauser, Ivens, Kummerfeld, Pereira-Leal, Nilsson, Peterson, Salzberg, Shallom, Silva, Sundaram, Westenberger, White, Melville, Donelson, Andersson, Stuart and Hall2005b; Porcel et al., Reference Porcel, Denoeud, Opperdoes, Noel, Madoui, Hammarton, Field, Da Silva, Couloux, Poulain, Katinka, Jabbari, Aury, Campbell, Cintron, Dickens, Docampo, Sturm, Koumandou, Fabre, Flegontov, Lukeš, Michaeli, Mottram, Szoor, Zilberstein, Bringaud, Wincker and Dollet2014; Kraeva et al., Reference Kraeva, Butenko, Hlaváčová, Kostygov, Myškova, Grybchuk, Leštinová, Votýpka, Volf, Opperdoes, Flegontov, Lukeš and Yurchenko2015; Flegontov et al., Reference Flegontov, Butenko, Firsov, Kraeva, Eliáš, Field, Filatov, Flegontova, Gerasimov, Hlaváčová, Ishemgulova, Jackson, Kelly, Kostygov, Logacheva, Maslov, Opperdoes, O'Reilly, Sádlová, Ševčíková, Venkatesh, Vlček, Volf, Votýpka, Záhonová, Yurchenko and Lukeš2016) reveals that the adoption of the parasitic lifestyle has led to a reduction in gene number approximately by half. Despite this dramatic reduction in gene number, B. saltans and Trypanosomatidae still share about 2800 homologous protein-coding genes. In this section we concentrate only on a core subset of 581 house-keeping genes involved in metabolism. We followed their losses and gains throughout trypanosomatid evolution, always using B. saltans as an outgroup. An interactive phylogenetic tree showing these gains and losses can be accessed at http://big.icp.ucl.ac.be/~opperd/metabolism/kinetoplastida_LGT4.html
Emergence of a parasite: the first steps
Iron is an essential element for all living organisms. In order to survive inside their hosts, parasites must gain access to their host's iron stores. Similar to disease-causing bacteria that release iron-binding molecules such as siderophores or scavenge iron from host haemoglobin and transferrin, parasites have developed mechanisms that allow them to compete for the limited amounts of free iron in the insect or mammalian host. A recent identification of a ferric iron reductase [LFR1 (Flannery et al., Reference Flannery, Huynh, Mittra, Mortara and Andrews2011)], a ferrous iron transporter [LIT1 (Jacques et al., Reference Jacques, Andrews and Huynh2010)], a haem transporter [LHR1 (Miguel et al., Reference Miguel, Flannery, Mittra and Andrews2013)] and the haem scavenging protein [LABCG5 (Flannery et al., Reference Flannery, Renberg and Andrews2013)] as virulence factors of Leishmania spp., has allowed us to identify the sequence of events involved in putting essential trypanosomatid iron-capture mechanisms in place. One of the primary adaptations required for a parasitic lifestyle must have been the acquisition of a high-affinity receptor/transporter for the capture and internalization of ferrous iron. This permits the effective competition for the limited amounts of free iron in the tissue fluids of the insect host. Although the free-living common ancestor of trypanosomatids was able to reduce insoluble ferric iron to soluble ferrous iron by a ferric reductase (present in most Kinetoplastea including Bodo), a ferrous transporter was likely lacking in this organism. Bodo saltans, which can be considered as a proxy of such an ancestor, does not have this transporter, apparently because of its bacteriotrophic lifestyle providing the flagellate with sufficient amount of reduced iron. The genome of the early branching P. confusum, or its direct ancestor, acquired a single copy gene of a plant-like ZIP-family ferrous iron transporter (Jacques et al., Reference Jacques, Andrews and Huynh2010; Flannery et al., Reference Flannery, Renberg and Andrews2013), and multicopy genes appeared subsequently in all other trypanosomatids. This must have been one of the first steps towards parasitism. A similar scenario holds for the capture of haem. While all Kinetoplastea, including B. saltans, possess a LABCG5 homologue to compensate for the lack of haem biosynthesis, a dedicated haem transporter such as LHR1 was acquired by P. confusum, or its immediate ancestor, so permitting survival inside an insect host. This LHR1 gene was secondarily lost in one of the two plant-dwelling haem-lacking phytomonads and in the African trypanosome, T. vivax (Flannery et al., Reference Flannery, Renberg and Andrews2013). T. brucei, a blood-dwelling parasite, captures iron and haem via, respectively, a transferrin receptor (ESAG6/ESAG7) (van Luenen et al., Reference van Luenen, Kieft, Mussmann, Engstler, ter Riet and Borst2005) and a haptoglobin–haemoglobin receptor (Vanhollebeke et al., Reference Vanhollebeke, De Muylder, Nielsen, Pays, Tebabi, Dieu, Raes, Moestrup and Pays2008). Both receptors seem to be specific adaptations to a life in the bloodstream, because in the procyclic insect stage of T. brucei these genes are not expressed, and haem is acquired only via the haem uptake protein TbHrg (Tb927.8.6010, (Horáková et al., Reference Horáková, Changmai, Vancová, Sobotka, Van Den Abbeele, Vanhollebeke and Lukeš2017)), an orthologue of the Leishmania LHR1 (LmjF24.2230) that shares only 24% identical residues.
Speciation by gene losses
The acquisition of the parasitic lifestyle by the common ancestor of all Trypanosomatidae was likely associated with a progressive loss of metabolic capacities. However, with no genomic information about an organism immediately ancestral to both Bodo and Trypanosomatidae available, loss of genes in the trypanosomatids and gene acquisition in Bodo are equally possible. Thus, one should err on the side of caution with a scenario that follows. It seems likely that almost immediately after the transition from the free-living kinetoplastid to the last common ancestor of all Trypanosomatidae, approximately 9500 genes were lost (Jackson et al., Reference Jackson, Otto, Aslett, Armstrong, Bringaud, Schlacht, Hartley, Sanders, Wastling, Dacks, Acosta-Serrano, Field, Ginger and Berriman2016; Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016). Although most of these genes were members of some large multigene families (exemplified by the GP46-like surface antigen with 391 copies in the genome of B. saltans) or encoded enigmatic ‘hypothetical proteins’, a smaller number of them (35 from 581 analysed) encoded metabolic enzymes.
Several complete metabolic pathways became redundant because the corresponding products could be acquired from the host. Typical examples of such metabolic losses in trypanosomatids are Lys catabolism and aerobic degradation of the aromatic amino acids Phe and Tyr (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016). Moreover, most of the genes for Trp degradation were lost when P. confusum branched off from the main trypanosomatid lineage (Skalický et al., Reference Skalický, Dobáková, Wheeler, Tesařová, Flegontov, Jirsová, Votýpka, Yurchenko, Ayala and Lukeš2017). The His catabolism, still present in B. saltans and P. confusum, also disappeared from most of the trypanosomatids, with a single exception of T. cruzi.
An important evolutionary event was the loss of hydroxyl-methyl-glutaryl-CoA (HMG-CoA) lyase and β-hydroxy-butyrate dehydrogenase genes in Leishmaniinae. These enzymes are essential for the conversion of Leu into ketone bodies (acetoacetate and beta-hydroxybutyrate). Thus, all Leishmaniinae including Leishmania spp. use Leu over acetate for biosynthesis of their sterols (Ginger et al., Reference Ginger, Chance, Sadler and Goad2001). Members of the same clade also lost the gene for trypanosome alternative oxidase. Finally, P. confusum, the earliest-branching trypanosomatid, is the only species sharing a ‘protist-type’ arginase gene with B. saltans. This arginase was subsequently lost by all other trypanosomatids, while only the Leishmaniinae re-acquired an entirely different arginase gene from fungi (Gaur et al., Reference Gaur, Roberts, Dalvi, Corraliza, Ullman and Wilson2007; Opperdoes and Michels, Reference Opperdoes and Michels2007).
Bodo saltans, P. confusum and members of the genera Leishmania, Crithidia and Leptomonas are all able to metabolize the branched amino acids Ile and Val, as well as Met and Thr into the TCA-cycle intermediate succinate (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016). In contrast, Trypanosoma, Phytomonas and Blechomonas spp. are unable to metabolize these amino acids, as they independently lost three genes of the methyl malonyl-CoA pathway: propionyl-CoA carboxylase, methyl-malonyl-CoA mutase and methyl-malonyl-CoA epimerase. In addition, they lost xylulokinase, which is required for the utilization of the pentose sugar xylulose.
The African trypanosomes (T. vivax, T. brucei and T. congolense) and Phytomonas spp. have adapted to the life in glucose-rich fluids – mammalian blood and plant juices, respectively. Trypanosomes are able to reversibly suppress mitochondrial oxidative phosphorylation in favour of a metabolism exclusively geared towards the consumption of glucose, while phytomonads have irreversibly lost their cytochromes (Opperdoes, Reference Opperdoes1987; Sanchez-Moreno et al., Reference Sanchez-Moreno, Lasztity, Coppens and Opperdoes1992). Such a high degree of specialization has led to convergent evolution between the African trypanosomes and Phytomonas spp., characterized by a parallel loss of numerous genes. The African trypanosomes have lost 35 metabolic genes, while phytomonads have lost more than twice as much (Porcel et al., Reference Porcel, Denoeud, Opperdoes, Noel, Madoui, Hammarton, Field, Da Silva, Couloux, Poulain, Katinka, Jabbari, Aury, Campbell, Cintron, Dickens, Docampo, Sturm, Koumandou, Fabre, Flegontov, Lukeš, Michaeli, Mottram, Szoor, Zilberstein, Bringaud, Wincker and Dollet2014). Of these, the two groups have 16 losses in common. Those are involved in the synthesis of phosphonolipids, Met and tetra-hydrofolate, long-chain polyunsaturated acids, as well as conversions of Glu into Pro, Asn into Asp, and Ser into Gly. In addition, genes for methyl-glyoxal detoxification, formation of HMG-CoA from acetyl CoA, trans-hydrogenation via D-lactate dehydrogenase, tetrahydrofolate synthesis, Cys synthesis, β-oxidation of fatty acids, metabolism of ascorbate and pentose sugars, ribulokinase, quinonoid di-hydro-pteridine reductase, ascorbate peroxidase, and old yellow enzyme were all lost in these two highly specialized clades.
Speciation by gene gains
In the course of evolution, trypanosomatid genomes were reshaped not only by losses of genes but also by gene duplications and acquisitions via horizontal gene transfer. Early on in their evolution, more than 18 metabolic genes were acquired, possibly simultaneously. These include genes involved in the cyclopropane-fatty-acyl-phospholipid formation, bromodomain factor 1 permitting an additional level of enzyme regulation, and the ferrous iron transporter allowing more efficient competition for the soluble iron within the host. A biopterin/folate/pteridin transporter was originally acquired by the common ancestor of B. saltans and trypanosomatids from one of three possibilities, a cyanobacterium, plant or algal organism (Klaus et al., Reference Klaus, Kunji, Bozzo, Noiriel, de la Garza, Basset, Ravanel, Rebeille, Gregory and Hanson2005; Opperdoes and Coombs, Reference Opperdoes and Coombs2007). It is a single copy gene in B. saltans, but in all trypanosomatids it expanded into a multi-gene family. The number of its copies per haploid genome varies from 2 in Blechomonas and 4 in P. confusum, to over 50 copies in C. fasciculata. These folate transporter arrays, along with the acquisition of pteridine reductase, underline the importance of an efficient salvage of pteridines and their subsequent metabolism in the parasitic lifestyle.
In general, evolution of trypanosomatids featured significantly more losses than acquisitions of metabolic genes. An exception to this rule is the subfamily Leishmaniinae, which acquired considerably more metabolic genes (23) than were lost (4). Acquisitions include 3 genes of the haem biosynthetic pathway – protoporphyrinogen oxidase, coprophyrinogen III oxidase and ferrochelatase (Ivens et al., Reference Ivens, Peacock, Worthey, Murphy, Aggarwal, Berriman, Sisk, Rajandream, Adlem, Aert, Anupama, Apostolou, Attipoe, Bason, Bauser, Beck, Beverley, Bianchettin, Borzym, Bothe, Bruschi, Collins, Cadag, Ciarloni, Clayton, Coulson, Cronin, Cruz, Davies, De Gaudenzi, Dobson, Duesterhoeft, Fazelina, Fosker, Frasch, Fraser, Fuchs, Gabel, Goble, Goffeau, Harris, Hertz-Fowler, Hilbert, Horn, Huang, Klages, Knights, Kube, Larke, Litvin, Lord, Louie, Marra, Masuy, Matthews, Michaeli, Mottram, Muller-Auer, Munden, Nelson, Norbertczak, Oliver, O’Neil, Pentony, Pohl, Price, Purnelle, Quail, Rabbinowitsch, Reinhardt, Rieger, Rinta, Robben, Robertson, Ruiz, Rutter, Saunders, Schafer, Schein, Schwartz, Seeger, Seyler, Sharp, Shin, Sivam, Squares, Squares, Tosato, Vogt, Volckaert, Wambutt, Warren, Wedler, Woodward, Zhou, Zimmermann, Smith, Blackwell, Stuart, Barrell and Myler2005; Opperdoes and Coombs, Reference Opperdoes and Coombs2007), three genes of the urea cycle – argininosuccinate synthase, argininosuccinate lyase and arginase and two more genes involved in glycosylation reactions.
Speciation of the genus Trypanosoma is characterized by the acquisition of phospholipase A1, GPI inositol deacylase 2 gene, and the loss of genes encoding chitinase, cyclopropane-fatty-acyl-phospholipid synthase, both the cytosolic and mitochondrial serine hydroxyl-methyl-transferase isoenzymes, as well as xanthine phosphoribosyl transferase. The newly acquired phospholipase A1, PLA(1) is clearly distinct from the lysosomal isoenzyme (Opperdoes and van Roy, Reference Opperdoes and van Roy1982; Richmond and Smith, Reference Richmond and Smith2007b). The former lipase is an orthologue of a bacterial extracellular phospholipase A1 that was most likely acquired from a horizontal gene transfer from Sodalis glossinidius, a bacterial endosymbiont of tsetse flies (Richmond and Smith, Reference Richmond and Smith2007a). Interestingly, a BLAST search revealed that PLA1 is an orthologue of the T. brucei ESAG1, which encodes a transmembrane protein located in the flagellar pocket (Nolan et al., Reference Nolan, Garcia-Salcedo, Geuskens, Salmon, Paturiaux-Hanocq, Pays, Terbadi, Pays, Black and Seed2002). In the bloodstream stage of the African trypanosomes, it probably functions as a phospholipase which captures fatty acids and phospholipids by scavenging the lysophosphatidylcholine present in a sub-millimolar concentration in the host plasma (Uttaro, Reference Uttaro2014). In T. cruzi, a similar, but non-homologous PLPA1 isoenzyme was proposed as a putative virulence factor (Belaunzarán et al., Reference Belaunzarán, Wilkowsky, Lammel, Gimenez, Bott, Barbieri and de Isola2013). Trypanosomes have also acquired proline racemase gene, which was implicated in B-cell polyclonal activation, immunosuppression, and evasion of the host defense by T. cruzi (Reina-San-Martin et al., Reference Reina-San-Martin, Degrave, Rougeot, Cosson, Chamond, Cordeiro-Da-Silva, Arala-Chaves, Coutinho and Minoprio2000). This gene was also retained in T. vivax, but lost in T. brucei and T. congolense (Caballero et al., Reference Caballero, Costa-Martins, Ferreira, JM, Serrano, Camargo, Buck, Minoprio and MM2015).
Blechomonas ayalai and Phytomonas spp. share the gene encoding isopropanol dehydrogenase (Molinas et al., Reference Molinas, Altabe, Opperdoes, Rider, Michels and Uttaro2003). However, it seems to be functional only in Phytomonas, since the Blechomonas homologue appears pseudogenized.
Blechomonas ayalai and trypanosomes metabolize Thr via the Thr dehydrogenase pathway, which apparently became enabled after the acquisition of an additional Thr dehydratase gene by a common ancestor of all trypanosomatids except Paratrypanosoma. This event introduced the possibility of choice between two alternative pathways for Thr degradation (Opperdoes and Coombs, Reference Opperdoes and Coombs2007) and eventually led to a differential loss of either the Thr dehydrogenase or the Thr dehydratase pathway. This has resulted in the dramatic differences in the way this amino acid is metabolized in Trypanosoma and Leishmania. (Opperdoes and Coombs, Reference Opperdoes and Coombs2007).
The common ancestor of Leishmaniinae gained novel genes involved in sucrose and pentose sugar metabolism, as well as the catalase. The latter was then selectively lost in members of the genus Leishmania, likely due to their dixenous life cycle (Kraeva et al., Reference Kraeva, Horáková, Kostygov, Kořený, Butenko, Yurchenko and Lukeš2017). More recent acquisitions, shared only by Crithidia and Leptomonas, are genes encoding diaminopimelate metabolizing enzymes, β-glucosidase, nitroalkane oxidase, phenolic acid dehydrogenase and glycerol dehydrogenase. Genes involved in the conversion of the typical bacterial diaminopemelic acid into Lys are present only in Crithidia spp. and L. pyrrhocoris, but are absent in Leishmania spp. and L. seymouri.
Finally, B. saltans is not capable of ubiquinone biosynthesis, while all trypanosomatids encode proteins constituting this essential pathway. The most parsimonious scenario suggests that the phagotrophic lifestyle of B. saltans, which allows it to extract necessary ubiquinone from bacteria, facilitated the loss of these genes (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016).
Endosymbionts of trypanosomatids
Intracellular bacteria of trypanosomatids were discovered at the beginning of the 20th century in the monoxenous fly parasite Strigomonas culicis (at that time Blastocrithidia culicis) (Novy et al., Reference Novy, MacNeal and Torrey1907). With the advent of electron microscopy bacteria were also found in several other species of monoxenous trypanosomatids (Newton and Horne, Reference Newton and Horne1957; Mundim et al., Reference Mundim, Roitman, Hermans and Kitajima1974; Fiorini et al., Reference Fiorini, Faria e Silva, Soares and Brazil1989; Motta et al., Reference Motta, Cava, Silva, Fiorini, Soares and Desouza1991b). Their nature was confirmed by early analyses of DNA (Marmur et al., Reference Marmur, Cahoon, Shimura and Vogel1963), 70S ribosomes (Zaitseva and Salikhov, Reference Zaitseva and Salikhov1972), as well as chloramphenicol sensitivity (Zaitseva and Salikhov, Reference Zaitseva and Salikhov1973).
The early-described trypanosomatids’ intracellular bacteria are closely related to each other and so are the hosts harbouring them (Fig. 1). This suggests that bacterial acquisition was a single event in this group, which was followed by the subsequent long-term coevolution between the partners (Faria e Silva et al., Reference Faria e Silva, Sole-Cava, Soares, Motta, Fiorini and de Souza1991; Du and Chang, Reference Du and Chang1994; Du et al., Reference Du, Maslov and Chang1994a, Reference Du, McLaughlin and Chang1994b; Hollar et al., Reference Hollar, Lukeš and Maslov1998). The bacteria were assigned to the new beta-proteobacterial genus Kinetoplastibacterium (formally, Candidatus Kinetoplastibacterium) within the family Alcaligenaceae (Du et al., Reference Du, McLaughlin and Chang1994b), whereas their hosts were eventually united in two related genera – Angomonas and Strigomonas (Teixeira et al., Reference Teixeira, Borghesan, Ferreira, Santos, Takata, Campaner, Nunes, Milder, de Souza and Camargo2011). Kentomonas, the third genus in this group, was discovered recently and all three genera were assigned to a new subfamily Strigomonadinae to emphasize their relationship and shared features associated with endosymbiosis (Votýpka et al., Reference Votýpka, Kostygov, Kraeva, Grybchuk-Ieremenko, Tesařová, Grybchuk, Lukeš and Yurchenko2014).
The bacterial endosymbionts were also recorded in aquatic leech-transmitted trypanosomes – Trypanosoma cobitis (Lewis and Ball, Reference Lewis and Ball1980) and T. fallisi (Martin and Desser, Reference Martin and Desser1990; Reference Martin and Desser1991). In contrast to Strigomonadinae possessing only one bacterium per cell, the trypanosomes bear multiple intracytoplasmic bacteria. Regretfully, these studies were restricted to electron microscopy, and neither the identity of the endosymbionts nor their relationships with the flagellate hosts were investigated further.
The last bacterium-trypanosomatid endosymbiosis documented to date, that of Pandoraea novymonadis (beta-proteobacteria: Burkholderiaceae) and Novymonas esmeraldas (Leishmaniinae), has been described recently (Kostygov et al., Reference Kostygov, Dobáková, Grybchuk-Ieremenko, Váhala, Maslov, Votýpka, Lukeš and Yurchenko2016). As in the trypanosomes, there are multiple bacteria per flagellate cell. Because none of the partners in this endosymbiotic system has close relatives involved in such a relationship suggested its independent and relatively recent origin (Fig. 1). Nevertheless, analysis of the P. novymonadis genome indicated that these symbiotic relationships are already well established (Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017). Compared to Strigomonadinae, this endosymbiotic system remains understudied. Unlike the former, the specific insect host of N. esmeraldas is not known, as it has been documented in South American true bugs and African biting midges (Kostygov et al., Reference Kostygov, Dobáková, Grybchuk-Ieremenko, Váhala, Maslov, Votýpka, Lukeš and Yurchenko2016). Thus, at the moment it is not possible to study the endosymbiont influence on the flagellate fitness in the insect using experimental infections.
Different viruses can also infect trypanosomatids and play an important role in their biology (Ives et al., Reference Ives, Ronet, Prevel, Ruzzante, Fuertes-Marraco, Schutz, Zangger, Revaz-Breton, Lye, Hickerson, Beverley, Acha-Orbea, Launois, Fasel and Masina2011; Grybchuk et al., Reference Grybchuk, Akopyants, Kostygov, Konovalovas, Lye, Dobson, Zangger, Fasel, Butenko, Frolov, Votýpka, d'Avila-Levy, Kulich, Moravcová, Plevka, Rogozin, Serva, Lukeš, Beverley and Yurchenko2018a). We refer readers to several recent reviews discussing this topic (Lukeš et al., Reference Lukeš, Butenko, Hashimi, Maslov, Votýpka and Yurchenko2018; Grybchuk et al., Reference Grybchuk, Kostygov, Macedo, d'Avila-Levy and Yurchenko2018b).
Interactions of trypanosomatids with their bacterial endosymbionts
The relationships of Strigomonadinae and Novymonas with their endosymbionts demonstrate many important differences, which are noticeable even on the morphological/ultrastructural level. While P. novymonadis cells are localized in vacuoles and preserve a well-developed peptidoglycan layer in the cell wall (Kostygov et al., Reference Kostygov, Dobáková, Grybchuk-Ieremenko, Váhala, Maslov, Votýpka, Lukeš and Yurchenko2016), Kinetoplastibacterium spp. are situated directly in the cytoplasm of the host cell and their peptidoglycan layer is reduced (Chang, Reference Chang1974; Soares and De Souza, Reference Soares and De Souza1988; Motta et al., Reference Motta, Cava, Silva, Fiorini, Soares and Desouza1991b). The absence of a vacuolar membrane around bacteria and their thinner (and thereby more permeable) cell wall in the latter case apparently facilitate an intense metabolic exchange with the host enabling a mutually beneficial division of labour in metabolic pathways. The relationships between P. novymonadis and N. esmeraldas appear to be more primitive: the host keeps endosymbionts in vacuoles, likely to exercise more tight control over them. Occasionally, the trypanosomatid digests bacteria using lysosomes, probably in order to regulate their number and consume their products (Kostygov et al., Reference Kostygov, Dobáková, Grybchuk-Ieremenko, Váhala, Maslov, Votýpka, Lukeš and Yurchenko2016). Strigomonadinae do not need to use such a crude method to control the number of their endosymbionts. Instead, they evolved a fine-tuned mechanism ensuring precise coordination between the division of the trypanosomatid cell and its single intracellular bacterium (Motta et al., Reference Motta, Catta-Preta, Schenkman, de Azevedo Martins, Miranda, de Souza and Elias2010; Brum et al., Reference Brum, Catta-Preta, de Souza, Schenkman, Elias and Motta2014; Catta-Preta et al., Reference Catta-Preta, Brum, da Silva, Zuma, Elias, de Souza, Schenkman and Motta2015).
As mentioned above, the main role of the bacterial endosymbionts is to supply the trypanosomatid hosts with essential nutrients. One of these is haem, which trypanosomatids are unable to synthesize, although it is indispensable for the production of numerous important enzymes, such as the cytochromes (Gill and Vogel, Reference Gill and Vogel1963; Chang et al., Reference Chang, Chang and Sassa1975; de Menezes and Roitman, Reference de Menezes and Roitman1991; Kořený et al., Reference Kořený, Oborník and Lukeš2013).
Typical trypanosomatids require many vitamins for their growth, such as riboflavin, pantothenic acid, pyridoxamine, folic acid, thiamine, nicotinic acid and biotin (Roitman et al., Reference Roitman, Roitman and de Azevedo1972). However, Strigomonadinae require only the last three of them, since the others are supplied by the endosymbionts (Mundim et al., Reference Mundim, Roitman, Hermans and Kitajima1974; Klein et al., Reference Klein, Alves, Serrano, Buck, Vasconcelos, Sagot, Teixeira, Camargo and Motta2013). Interestingly, they perform all steps of the panthotenic acid synthesis, but the last one, which is completed by the flagellate host, demonstrating an intimate cooperation between the two partners (Klein et al., Reference Klein, Alves, Serrano, Buck, Vasconcelos, Sagot, Teixeira, Camargo and Motta2013). Pandoraea novymonadis is able to synthesize all the above-mentioned vitamins, thereby making its host, N. esmeraldas, not dependent on their availability in the environment (Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017). As for the amino acids, most trypanosomatids are unable to synthesize Arg, His, Ile, Leu, Phe, Trp and Tyr (Opperdoes et al., Reference Opperdoes, Butenko, Flegontov, Yurchenko and Lukeš2016). The same holds true for aposymbiotic strains of Strigomonadinae, which additionally require Cys, Lys, Met and Thr (Mundim and Roitman, Reference Mundim and Roitman1977; Freymuller and Camargo, Reference Freymuller and Camargo1981). Meanwhile, wild-type strains are auxotrophic only for Met and Tyr, which they apparently obtain from their insect hosts (Mundim et al., Reference Mundim, Roitman, Hermans and Kitajima1974; Alves et al., Reference Alves, Klein, da Silva, Costa-Martins, Serrano, Buck, Vasconcelos, Sagot, Teixeira, Motta and Camargo2013a). Owing to multiple horizontal gene transfers, synthetic pathways for several amino acids are interlaced between Kinetoplastibacterium spp. and their hosts, so that the enzymes missing in the bacteria are present in the trypanosomatids and vice versa, providing another example of their deep metabolic integration (Alves et al., Reference Alves, Klein, da Silva, Costa-Martins, Serrano, Buck, Vasconcelos, Sagot, Teixeira, Motta and Camargo2013a; Alves, Reference Alves and D'Mello2017). Pandoraea novymonadis is unable to synthesize Ala, Asn, Asp, Cys, Met and Pro; yet these can be synthesized by the flagellate. In return, the bacterium preserves the enzymes required for synthesis of nine amino acids, for which its host is auxotrophic (Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017).
The endosymbiotic lifestyle led to a significant genomic reduction of the intracellular bacteria in question (Alves et al., Reference Alves, Klein, da Silva, Costa-Martins, Serrano, Buck, Vasconcelos, Sagot, Teixeira, Motta and Camargo2013a, Reference Alves, Serrano, Maia da Silva, Voegtly, Matveyev, Teixeira, Camargo and Buck2013b; Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017). Besides the biosynthesis of amino acids, this reduction affected enzymes involved in the production of polyamines, which are essential for many cellular processes. Both Kinetoplastibacterium spp. and P. novymonadis rely on their hosts in this respect (Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017). It was demonstrated that the bacterial endosymbiont of Angomonas deanei enhances the activity of host's ornithine decarboxylase, which leads to an intensification of the polyamine synthesis and accelerated the proliferation of the trypanosomatid (Frossard et al., Reference Frossard, Seabra, DaMatta, de Souza, de Mello and Motta2006). Since Kinetoplastibacterium spp. lost their ability to synthesize important components of membranes such as cardiolipin, phosphatidylethanolamine, and phosphatidylserine, these phospholipids have to be supplied by the flagellate hosts. In contrast, P. novymonadis is self-dependent in this regard, consistently with its more secluded mode of life within the host cell.
The metabolism of Strigomonadinae (glycolysis, ATP production and hydrolysis, oxygen consumption and oxidation–reduction processes) was shown to be boosted in the presence of the endosymbiont (Penha et al., Reference Penha, Hoffmann, Souza, Martins, Bottaro, Prosdocimi, Faffe, Motta, Urmenyi and Silva2016; Loyola-Machado et al., Reference Loyola-Machado, Azevedo-Martins, Catta-Preta, de Souza, Galina and Motta2017). While in the aposymbiotic Strigomonadinae the glycosomes are dispersed throughout the cytoplasm, in the symbiont-containing cells they are closely associated with the bacteria providing direct access to ATP (Motta et al., Reference Motta, Soares, Attias, Morgado, Lemos, Saad-Nehme, Meyer-Fernandes and De Souza1997; Faria-e-Silva et al., Reference Faria-e-Silva, Attias and de Souza2000; Loyola-Machado et al., Reference Loyola-Machado, Azevedo-Martins, Catta-Preta, de Souza, Galina and Motta2017). The mitochondrion of Strigomonadinae demonstrates an extensive branching on the periphery of the cell, with the consequent reorganization of subpellicular microtubules (Freymuller and Camargo, Reference Freymuller and Camargo1981). In N. esmeraldas, the single mitochondrion also seems to be hypertrophied, although its peripheral projections do not distort the microtubular corset (Kostygov et al., Reference Kostygov, Dobáková, Grybchuk-Ieremenko, Váhala, Maslov, Votýpka, Lukeš and Yurchenko2016). Combined, these findings suggest an enhanced energy consumption of endosymbiont-containing trypanosomatids.
The Kinetoplastibacterium spp. affect the surface charge and composition of glucoconjugates on the trypanosomatid plasma membrane (Dwyer and Chang, Reference Dwyer and Chang1976; Esteves et al., Reference Esteves, Andrade, Angluster, de Souza, Mundim, Roitman and Perreira1982; Motta et al., Reference Motta, Saraiva, Costa e Silva Filho and de Souza1991a; de Faria-e-Silva et al., Reference de Faria-e-Silva, Costa e Silva-Filho and de Souza1999), which are responsible for the different efficiency of infecting the insect host documented for wild-type and aposymbiotic strains (Fampa et al., Reference Fampa, Correa-da-Silva, Lima, Oliveira, Motta and Saraiva2003; d'Avila-Levy et al., Reference d'Avila-Levy, Silva, Hayashi, Vermelho, Alviano, Saraiva, Branquinha and Santos2005). Moreover, this also correlates with the activities of ecto-phosphatases and gp63-like proteases differing in the endosymbiont-bearing and bacteria-free trypanosomatids (d'Avila-Levy et al., Reference d'Avila-Levy, Santos, Marinho, Matteoli, Lopes, Motta, Santos and Branquinha2008; Catta-Preta et al., Reference Catta-Preta, Nascimento, Garcia, Saraiva, Motta and Meyer-Fernandes2013).
The intimate and complex interactions between the cellular processes of the bacterial endosymbionts and their trypanosomatid hosts require a well-developed signalling system. Indeed, it has been demonstrated that the outer membrane of Strigomonadinae contains phosphatidylcholine, a host-produced lipid participating in cell signalling, which is typical for eukaryotes and their symbionts (Palmié-Peixoto et al., Reference Palmié-Peixoto, Rocha, Urbina, de Souza, Einicker-Lamas and Motta2006; de Azevedo-Martins et al., Reference de Azevedo-Martins, Frossard, de Souza, Einicker-Lamas and Motta2007; Reference de Azevedo-Martins, Alves, de Mello, Vasconcelos, de Souza, Einicker-Lamas and Motta2015).
It still remains puzzling why a few groups of trypanosomatids evolved to compensate for their deficiency in synthetic capabilities by acquiring endosymbionts, while the majority remain restricted to nutrients supplied by their insect hosts. This may be related to the differences in the life cycles, which are largely unknown for the majority of monoxenous trypanosomatids.
Conclusions and perspectives
The recent years were characterized by several significant advances in the field of trypanosomatid biology. Technologically, this progress was dynamically driven forward by a wide-scale application of genomics (and the related -omics) tools, as well as further improvements in biochemical, reverse genetics, and microscopy approaches. The recent advances include (but are not limited to) new insights in trypanosomatid genetics and sexual processes, biodiversity and population structure, virulence factors and other aspects of host–parasite interactions, transitions from monoxenous to dixenous lifestyle, epigenetics and its role in VSG switching, enzymology of RNA editing, and studies of associated microbiota. Yet, many unanswered and exciting questions still remain awaiting new ideas, unorthodox experimental approaches, and perhaps, a new generation of scientist to tackle them.
Acknowledgements
We thank the members of our laboratories for fruitful discussions.
Financial support
The support from the Grant Agency of Czech Republic awards 16-18699S (JL and VY), 17-10656S (VY), 17-24036S (HH), 18-15962S (VY and JL), the COST action CM1307 (FRO and JL), the European Research Council CZ LL1601 (JL), and the project ‘Centre for Research of Pathogenicity and Virulence of Parasites’ (No. CZ.02.1.01/0.0/0.0/16_019/0000759) funded by the European Regional Development Fund and Ministry of Education, Youth and Sports of the Czech Republic (JL, HH, AYK and VY) is thankfully acknowledged. The funders had no role in data collection, decision to publish or preparation of the manuscript.
Conflicts of interest
None.
Ethical standards
Not applicable.

