


Introduction
Leishmaniasis, with three basic clinical manifestations of the disease: cutaneous (CL), mucocutaneous (MCL) and visceral leishmaniasis (VL) caused by over 15 different species of the intracellular protozoan parasite Leishmania. With an estimated 12 million cases of leishmaniasis worldwide and 1.5–2 million new cases reported each year (Herwaldt, Reference Herwaldt1999). The parasite is primarily found in South America, Asia, southern Europe and Africa (Guerin et al. Reference Guerin, Olliaro, Sundar, Boelaert, Croft, Desjeux, Wasunna and Bryceson2002). Since there is no antileishmanial vaccine is clinically available, control of Leishmaniasis relies almost exclusively on chemotherapy. For the past 7 decades pentavalent antimonials constituted the standard antileishmanial treatment worldwide, however, the last 20 years their clinical value was imperiled due to the widespread emergence of resistance to these agents in Bihar, India, where more than 50% of VL cases occur globally (Sundar et al. Reference Sundar, Chakravarty, Rai, Agrawal, Singh, Chauhan and Murray2007). The availability of molecular methods, genome sequence, proteomic methods to the study Leishmania biology is practical and quite possible. Researchers should have to apply these methods and effective tools to the study of resistance mechanisms/novel drug targets and speed up the process of antileishmanial drug discovery.
Laboratory-induced drug resistance and field isolate resistance mechanism is partly understood in Leishmania. The high-level resistance to antimony observed in Leishmania can be due to decreased drug uptake and simultaneous selection of loss in metal reduction (Faraut-Gambarelli et al. Reference Faraut-Gambarelli, Piarroux, Deniau, Giusiano, Marty, Michel, Faugere and Dumon1997; Rojas et al. Reference Rojas, Valderrama, Valderrama, Varona, Ouellette and Saravia2006). In contrast to in vitro selected strains, resistance to Sb(V) in Leishmania field isolates is not well understood (Carrio et al. Reference Carrio, Riera, Gallego, Ribera and Portus2001; Hadighi et al. Reference Hadighi, Mohebali, Boucher, Hajjaran, Khamesipour and Ouellette2006). Other available antileishmanial drugs are also showing resistance in different part of the world and resistance mechanism is unclear. Current studies with field isolates of Leishmania (Lira et al. Reference Lira, Sundar, Makharia, Kenney, Gam, Saraiva and Sacks1999; Kumar et al. Reference Kumar, Sisodia, Misra, Sundar, Shasany and Dube2010) suggest similarities and differences with laboratory-resistant isolates but it is not yet clear whether any of the highlighted mechanisms is central to the resistance phenotype of field isolates. Understanding how antileishmanial drugs work and why they sometimes fail is fundamental to the optimal use of the existing formulations and is also likely to be instructive in the development of new therapies. It, therefore, appears quite clear that the cadence of antileishmanial drug development has not kept pace with the clinical needs. In this review, we further discuss the antileishmanial drug developed in past decades, current scenario of those drugs, emerging/novel drug targets, and challenges of antileishmanial drug development.
Journey of antileishmanial drugs development
The therapeutic options for Leishmania infections are quite limited and include only a few drugs (Fig. 1). The timeline of these drug discoveries or uses is explained in Fig. 2 that indicated the slow pace of antileishmanial discovery. Only one new class of antileishmanial drugs in terms of oral formulation has been developed in the last 20–30 years but they are also showing resistance now a day. Route of administration, mechanism of action and side effect of available antileishmanials are summarized and discussed in Table 1. Due to unavailability of an effective vaccine against leishmaniasis, drugs are only the important available means for the treatment.

Fig. 1. Chemical structure of few drugs used as therapeutic options for Leishmania infections. Sodium stibogluconate is first line antileishmanials. Pentamidine, Amphotericin B and Parmomycin are second-line drugs, exhibited to be effective against CL and VL. Miltefosine, allopurinol and sitamaquine have shown oral activity against Leishmaniasis and are in phase III/IV clinical trial.

Fig. 2. The timeline of antileishmanial drug discoveries from early 1920 to 2017, indicating the slow pace of antileishmanial discovery.
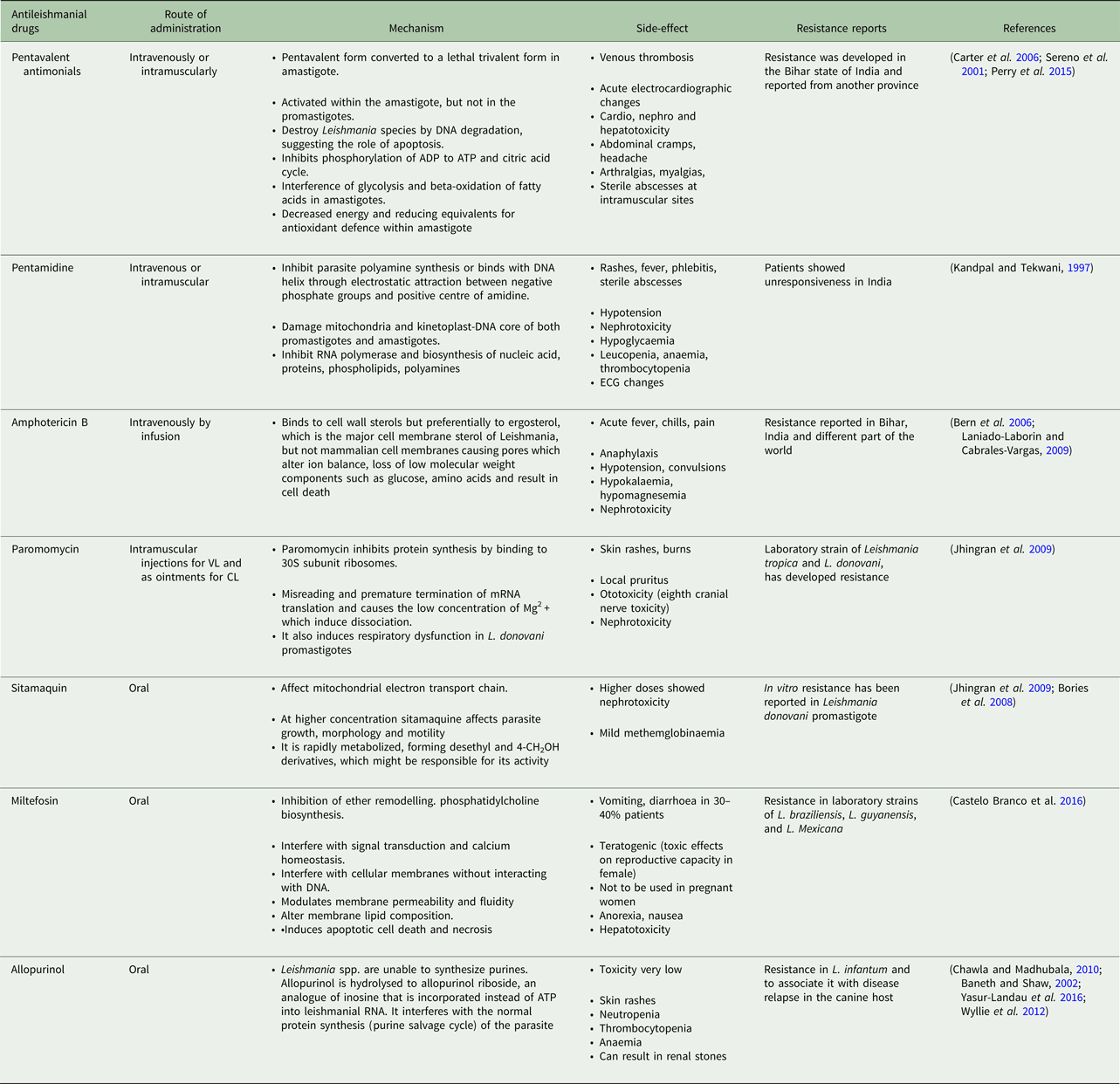
Table 1. Current antileishmanial drugs and their associated information
Preliminary/initial antileishmanials
Urea stibamine (Urea salt of Stibnic acid) was used as a first effective drug against Leishmania donovani in 1920. It saved thousands of lives in India from 1920 to 1930. After that potassium antimony tartrate (Tarter emetic) was started to use against Leishmania in Vianna. Stibamine and stibacetin were also used for the treatment of Leishmania but showed few side-effects. Then pentavalent antimonials were developed as the first line of drug for the treatment of Leishmaniasis with reduced side- effects at that time.
First-line antileishmanials
The pentavalent antimonials were first introduced in 1945 and remained the standard drugs for about 6 decades. The two compounds currently available for clinical use are sodium stibogluconate [(SSG) also known as sodium antimony gluconate (SAG) and its trade name is Stibanate/Pentostam)] and Meglumine antimoniate (also known as N-methylglucamine antimoniate and its trade name is Glucantime/Prostib). Stibogluconate {Sb(V)} under the brand of Pentostam has been manufactured by GlaxoWellcome which contains SSG. Using intracellular susceptibility testing, parasites isolated from Sb(V) unresponsive patients were found to be more resistant to Sb(V) as compared with parasites isolated from Sb(V) responsive patients (Kumar et al. Reference Kumar, Boggula, Sundar, Shasany and Dube2009). Several side-effects have decreased the use of antimonials.
Second-line antileishmanials
The methanesulphonate or isethionate salts of pentamidine have been used as second-line drugs in the treatment of VL refractory to antimonial treatment. However, the effectiveness of pentamidine is rapidly declining in India, suggesting that Leishmania parasites are becoming resistant (Sundar, Reference Sundar2001). Amphotericin B (polyene macrolide antifungal antibiotic) was discovered in the 1960s, from a bacterium of genus Streptomyces and given to patients who are failed to treat with SSG/SAG or pentamidine. More effective lipid-associated formulations of Amphotericin B are also available. Its instability at high temperature, painful delivery and high cost limits its usefulness. Parmomycin (aminoglycoside antibiotic) another second-line drug, exhibited to be effective against CL in clinical studies in Russia, and in clinical trials with VL in the early 1990s. Paromomycin in combination with stibogluconate was used successfully for treating SSG unresponsive VL in India and Kenya (Moore and Lockwood, Reference Moore and Lockwood2010).
Oral antileishmanials
Few agents (miltefosine, allopurinol, sitamaquine) have shown oral activity against Leishmaniasis. They are in phase III/IV clinical trial. Miltefosine was the first oral drug for VL. It is originally developed as an anticancer agent for topical treatment of skin metastases in 1992 but started to use for VL after 2000. Its activity against L. donovani was discovered in 1987 (Croft et al. Reference Croft, Neal, Pendergast and Chan1987). Allopurinol (Hypoxanthine analogue) was used for CL in 1991 first time and its efficacy against Indian Kala-azar was first reported (Saenz et al. Reference Saenz, Paz, Johnson, Marr, Nelson, Pattishall and Rogers1989). Allopurinol is ineffective as monotherapy against VL or CL. Now it is used with SSG for combination therapy. Sitamaquine (8-aminoquinoline and primaquine analogue) was developed by Walter Reed Army Institute of Research, the USA in 1994 for treatment. It is under development for over 8 years by Glaxo Smithkline, UK and Walter Reed Army Institute of Research, USA but the progress is very slow.
Current scenario of available drugs
Pentavalent antimonials
Despite the concern of proliferative resistance and various side-effects, pentavalent antimonials (SbV), the generic SSG (pentosam), N-methyl-glucamine antimoniate (Glucantime) and branded meglumine antimoniate, etc. are being used as a drug of choice for the treatment of Leishmaniasis over more than 5 decades (Singh et al. Reference Singh, Pandey and Sundar2006), but during the last two decades, antimonial resistance and therapeutic failures reached epidemic dimensions. Inadequate treatment in terms of dosing and duration and poor compliance promote the widespread antimonial resistance in an endemic area (Maltezou, Reference Maltezou2010). Pentavalent antimonials (SbV) considered as a prodrug, which should convert to trivalent antimonials (SbIII) in order to demonstrate their antileishmanial activity (Carter et al. Reference Carter, Hutchison, Henriquez, Legare, Ouellette, Roberts and Mullen2006). The reduction of pentavalent to trivalent compound takes place either in macrophages or in the parasite, however, it is still a dilemma (Shaked-Mishan et al. Reference Shaked-Mishan, Ulrich, Ephros and Zilberstein2001). Parasite-mediated reduction has been found to be associated with the loss of reductase activity of parasite, which may also lead to drug resistance. Though the exact mechanism of action is unknown (Sereno et al. Reference Sereno, Holzmuller, Mangot, Cuny, Ouaissi and Lemesre2001), but both forms (SbIII and SbV) of antimonials destroy Leishmania species by DNA degradation, suggesting the role of apoptosis, adenosine diphosphate phosphorylation and oxidation of fatty acid. Furthermore, the antimonials are also concerned with increasing the efflux of intracellular thiols by multi-drug-resistant protein A (MRPA), glycolysis inhibition and ABC (ATP-binding cassette) transporters (El Fadili et al. Reference El Fadili, Messier, Leprohon, Roy, Guimond, Trudel, Saravia, Papadopoulou, Legare and Ouellette2005). In natural antimonial resistance, the impaired thiol metabolism results in inhibition of SbV activation and decreased uptake of the active form SbIII by amastigotes; this is accomplished by the lower expression aquaglyceroporin 1 gene, which is involved in the uptake of SbIII (Decuypere et al. Reference Decuypere, Rijal, Yardley, De Doncker, Laurent, Khanal, Chappuis and Dujardin2005). The major cause of acquired resistance was the mismanagement of drug (Perry et al. Reference Perry, Prajapati, Menten, Raab, Feldmann, Chakraborti, Sundar, Fairlamb, Boelaert and Picado2015), due to its easy availability in its endemic regions along with the loss of drug activation by parasites. The transporters of pentamidine-resistant protein 1(PRP1) MRPA and ABC family that act as an efflux pump for antimonials, are also associated with antimony resistance (Coelho et al. Reference Coelho, Beverley and Cotrim2003).
Pentamidine
A drug which can be used as a second-line drug against Leishmaniasis is pentamidine, an aromatic diamine. It contains two main salts methansulphonate and isothionatethatare mainly used for the treatment of VL. In India, initially it was used to treat SbV refractory patients, but due to its drawbacks of decreased efficiency and high resistance, it was no longer be used. Later on, this drug was also used in combination with other drugs. In a study, patients which were unresponsive to antimony revealed better response for a combination of a low dosage of pentamidine and allopurinol as compare to the full dosages of pentamidine (Das et al. Reference Das, Ranjan, Sinha, Verma, Lal, Gupta, Siddiqui and Kar2001). Furthermore, a comparative study in Peru onmeglumine antimoniate (glucantime) and pentamidine against CL due to Leishmania braziliensis shows that glucantime was much more operational than pentamidine (Andersen et al. Reference Andersen, Cruz-Saldarriaga, Llanos-Cuentas, Luz-Cjuno, Echevarria, Miranda-Verastegui, Colina and Berman2005). Although, its precise mode of action is not known, in a study (Kandpal and Tekwani, Reference Kandpal and Tekwani1997) it is reported that in L. donovani promastigote the drug enters through polyamine and arginine transporters. Pentamidine is a highly toxic drug causes serious side-effects such as nephrotoxicity, hypoglycaemia and hypotension, etc. Although the mechanism of pentamidine resistance is not very well understood, ABC protein PRP1 can be associated with the resistance of pentamidine in the intracellular stage of Leishmania (Coelho et al. Reference Coelho, Messier, Ouellette and Cotrim2007).
Amphotericin B
Another drug which was previously being reported against Lieshmanial infection is Amphotericin B (AmB), a macrolide polyene antifungal drug widely used to treat systemic fungal infections(Marcondes et al. Reference Marcondes, Biondo, Gomes, Silva, Vieira, Camacho, Quinn and Chandrashekar2011). AmB is the drug of choice in the endemic areas of Bihar, India where antimonials resistance is common (Bern et al. Reference Bern, Adler-Moore, Berenguer, Boelaert, den Boer, Davidson, Figueras, Gradoni, Kafetzis, Ritmeijer, Rosenthal, Royce, Russo, Sundar and Alvar2006). AmB binds to membrane ergosterol, the predominant sterol of the fungal and leishmanial cell membrane. Even though it is highly effective but AmB also shows toxicity and its side-effects have been reported (Laniado-Laborin and Cabrales-Vargas, Reference Laniado-Laborin and Cabrales-Vargas2009). With its three clinical formulations, Ominous effects of plain AmB have been circumvented in which deoxycholate have been replaced by other lipids. These formulations are AmB colloidal dispersion (ABCD: Amphocil), liposomal AmB (L-AmB: Ambiosome) and AmB lipid complex (ABL: Abelcit). These lipid formulations of AmB retain their antifungal activity and show very high efficacy to cure this fatal disease and are less toxic. In VL cases, liposomal AmB has been proved as an efficient drug but its high cost limits its use to a common man suffering from this lethal disease. In a study, it was found that the success of AmB treatment mainly depends on immunity status of the patient and indicates that consecutive relapse could increase exposure of AmB resistant isolates (Di Giorgio et al. Reference Di Giorgio, Faraut-Gambarelli, Imbert, Minodier, Gasquet and Dumon1999). Some investigations have shown that resistance to AmB was found to be associated with gene TarII 64.4 and TarII 512.2 amplification in L. tarentolae mutant cell lines (Singh et al. Reference Singh, Papadopoulou and Ouellette2001) and overexpression of L-asparaginase gene in L. donovani (Singh et al. Reference Singh, Khan, Singh Yadav, Srivastava, Sinha, Ashish Das and Kundu2017). This finding warrants the possibility of resistance against the most successful drug.
Paromomycin
Paromomycin is broad-spectrum antibiotic produced by the bacterial spp. Streptomyces riomosus was originally developed as an oral drug for the treatment of intestinal protozoans in the 1960s. Later on, in 2002, it was first introduced as an antileishmanial drug in the form of PM sulphate (Williams et al. Reference Williams, Mullen, Baillie and Carter1998). Due to its low cost, better efficacy, shorter duration of administration and fewer side-effects it became very popular and thought to be a candidate for the first line therapy for VL patients. However, VL and CL both can be cured by paromomycin but its finite accessibility limited its use in endemic regions (Thakur, Reference Thakur2003). Due to its limited use resistance is not yet recorded in patient treatment but reported in vitro in L. tropica and L. donovani (Jhingran et al. Reference Jhingran, Chawla, Saxena, Barrett and Madhubala2009). To overcome these problems PM is being reported in combination with liposome and albumin microsphere and thereby exhibiting better results (Wiwanitkit, Reference Wiwanitkit2012).
Sitamaquine
Sitamaquine was developed for the treatment of VL, chemically known as 4-methyl-6-methoxy-8-aminoquinoline. The main foredeal of this drug is its oral administration. The Kenyan phase II trial of sitamaquine gave an opinion about its efficacy, against VL and was well endured (Wasunna et al. Reference Wasunna, Rashid, Mbui, Kirigi, Kinoti, Lodenyo, Felton, Sabin, Albert and Horton2005). However, despite its efficacy, some side-effects such as a headache, abdominal pain and kidney dysfunctioning were observed. The repercussion of Indian phase II trial was different from Kenyan trial (Jha et al. Reference Jha, Sundar, Thakur, Felton, Sabin and Horton2005). The Indian trial showed somewhat equal efficacy but the side-effects observed were vomiting, dyspepsia, cyanosis, nephritic syndrome and glomerulonephritis. At higher concentration sitamaquine affects parasite growth, morphology and motility (Duenas-Romero et al. Reference Duenas-Romero, Loiseau and Saint-Pierre-Chazalet2007). The mechanism involves electrostatic interaction between positively charged sitamaquine and phospholipid anionic polar head group and the insertion of the drug within the biological membrane is facilitated by phospholipid acyl chain (Coimbra et al. Reference Coimbra, Goncalves-da-Costa, Costa, Giarola, Rezende-Soares, Fessel, Ferreira, Souza, Abreu-Silva and Vasconcelos2008). In clinical practices, the resistance against this drug has not been reported yet but in vitro resistance has been reported against L. donovani promastigote by selecting the drug pressure of sitamaquine at the concentration of 160 µm (Bories et al. Reference Bories, Cojean, Huteau and Loiseau2008). Due to less activity and efficacy, its further clinical trials were restricted seriously (Garnier et al. Reference Garnier, Brown, Lawrence and Croft2006).
Miltefosine
The recently introduced drug for the antilieshmanial activity is miltefosine, an alkylphosphocholine also known as hexadecylphosphocholine which was originally developed as anticancerous agent (Croft and Coombs, Reference Croft and Coombs2003). Being used as a first oral drug for the treatment of Indian VL it was considered a major breakthrough in antileishmanial chemotherapy (Sundar et al. Reference Sundar, Jha, Thakur, Bhattacharya and Rai2006). Most of the studies (Sundar et al. Reference Sundar, Makharia, More, Agrawal, Voss, Fischer, Bachmann and Murray2000) shown that >95% of patients were cured regularly at this dosing pattern. Later on, it was observed that few patients were falling back again after 9–12 months of successful treatment with miltefosine. After that this drug was also used in combination with ambisome and found working effectively, but some of its side-effects (Castelo Branco et al. Reference Castelo Branco, Soares, de Jesus, Moreira, Alves, de Castro Belfort, Silva and Ferreira Pereira2016) raised questions about the effectiveness of this combination drug (Sundar et al. Reference Sundar, Sinha, Rai, Verma, Nawin, Alam, Chakravarty, Vaillant, Verma, Pandey, Kumari, Lal, Arora, Sharma, Ellis, Strub-Wourgaft, Balasegaram, Olliaro, Das and Modabber2011). The clinical resistance is not yet clear but being an oral drug it was used improperly in endemic countries like India, that became the reason for its resistance. It is reported that single point mutation at LdRos3 and LdMT or overexpression of multidrug-resistant gene MDR1 rendered the parasites remarkably less sensitive to miltefosine, and this resistance persisted in vivo; cross-resistance with other antileishmanial was not detected (Perez-Victoria et al. Reference Perez-Victoria, Parodi-Talice, Torres, Gamarro and Castanys2001; Pérez-Victoria et al. Reference Pérez-Victoria, Sánchez-Cañete, Seifert, Croft, Sundar, Castanys and Gamarro2006; Seifert et al. Reference Seifert, Pérez-Victoria, Stettler, Sánchez-Cañete, Castanys, Gamarro and Croft2007). In all miltefosine resistant Leishmania lines decreased drug accumulation is the common phenomenon, and this may be due to decreased uptake, increased efflux, faster metabolism or altered plasma membrane permeability; the first two mechanisms have been described in experimental models of miltefosine resistance (Pérez-Victoria et al. Reference Pérez-Victoria, Sánchez-Cañete, Seifert, Croft, Sundar, Castanys and Gamarro2006; Seifert et al. Reference Seifert, Pérez-Victoria, Stettler, Sánchez-Cañete, Castanys, Gamarro and Croft2007). Further, its long half-life and teratogenic properties (Sundar and Olliaro, Reference Sundar and Olliaro2007) are the two major drawbacks that became the reason of its clinical resistance and eventually lead to the end of its use as a regular drug.
Allopurinol
Allopurinol is second line drug used for the treatment of leishmaniasis due to unresponsiveness or resistance of first-line antileishmanial drugs. The antileishmanial activity of allopurinol was first reported in 1974 (Pfaller and Marr, Reference Pfaller and Marr1974). It is an analogue of purine and administered by oral route. Allopurinol is used alone or in combination with other drugs to the control of infection. Metabolization of allopurinol results into interruption of the pathogen's protein synthesis process, as they are not able to synthesize purines (Chawla and Madhubala, Reference Chawla and Madhubala2010). Specifically, it is attributed to the inhibition of the leishmanial enzyme hypoxanthine-guanine phosphoribosyl transferase (HGPRT). HGPRT takes part in the purine salvage pathway of the parasite. It converts dephosphorylated purines to nucleoside monophosphate leading to disrupted protein translation as well as the death of the parasite. Leishmaniasis has been treated for many years with allopurinol in domestic dogs and humans (Baneth and Shaw, Reference Baneth and Shaw2002). Recently some researchers have reported resistance of allopurinol in L. infantum and associated it with disease relapse in the canine host dog because the dog is the main reservoir for this zoonotic infection (Yasur-Landau et al. Reference Yasur-Landau, Jaffe, David and Baneth2016).
Beside above mentioned antileishmanial drugs few other compounds also been shown antiLeishmania potency, e.g. the anti-trypanosome drug fexinidazole showed potential for treating VL (Wyllie et al. Reference Wyllie, Patterson, Stojanovski, Simeons, Norval, Kime, Read and Fairlamb2012), Nitroimidazo–oxazole compound DNDI-VL-2098: an orally effective preclinical drug candidate (Gupta et al. Reference Gupta, Yardley, Vishwakarma, Shivahare, Sharma, Launay, Martin and Puri2014) and the R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for VL (Patterson et al. Reference Patterson, Wyllie, Stojanovski, Perry, Simeons, Norval, Osuna-Cabello, De Rycker, Read and Fairlamb2013). The main point of concern of antileishmanial drug discovery is the stepwise increase in resistance. The current scenario is worst and research going on Leishmania cannot rule out about the future availability of the candidate leishmanial antigens/immunogens. So further it needs extensive study and validation of emerging potent and novel drug targets of Leishmania.
Emerging and novel drug targets in Leishmania
Discovery of new potential drug targets mainly depends on biochemical, metabolic pathways and their enzymes, which are substantive for the survival of the parasite. Particular enzymes which are present in the parasite and absent from mammalian cells are of great interest. However, still, mammalian enzymes, which have structural homology, might also provide objectives for antileishmanial therapy because report showed favorable interactions of inhibitors with the active pocket of parasite enzyme, without adversely affecting human enzyme (Singh et al. Reference Singh, Khan, Singh Yadav, Srivastava, Sinha, Ashish Das and Kundu2017) and this is the rationale of the listed potential target in Leishmania. They could be interesting and promising for future antileishmanial drug development.
Enzymes of Trypanothione synthesis pathway
Trypanothione (bis-glutathionyl spermidine) is an indispensable molecule, which protects Leishmania against oxidative stress. Two enzymes namely trypanothione reductase (TR) and trypanothione synthetase (TS) catalyse the synthesis of trypanothione. TS catalyses the synthesis of trypanothione from two molecules of spermidine and glutathione, then in the presence of NADPH, trypanothione is upheld in its reduced form by the enzyme TR (Fairlamb et al. Reference Fairlamb, Blackburn, Ulrich, Chait and Cerami1985). Reduced trypanothione, in turn, reduces tryparedoxin (TX) followed by reduction of TX recycling enzyme tryparedoxin peroxidase (TP). Since, in Leishmania, this is the only pathway that is decisively implicated in maintaining oxidative stress, therefore, both TP and TR have been shown to be important drug targets. Null mutation of TR show attenuated infectivity (Dumas et al. Reference Dumas, Ouellette, Tovar, Cunningham, Fairlamb, Tamar, Olivier and Papadopoulou1997). The role of TR as potential targets for rational drug design has also been reviewed recently (Kumar et al. Reference Kumar, Ali and Bawa2014). Thus, enzymes of Trypanothione synthesis pathway provide a promising drug target in trypanosomatids.
Metacaspases
Another kind of molecule which can be used as a drug target in Leishmania is metacaspase which is believed to cause apoptosis (Lee et al. Reference Lee, Gannavaram, Selvapandiyan and Debrabant2007). Leishmania major metacaspase has been found to be essential for proper segregation of nucleus and kinetoplast of the parasite (Denise et al. Reference Denise, Poot, Jimenez, Ambit, Herrmann, Vermeulen, Coombs and Mottram2006). It is reported that under peroxide stress, metacaspase causes apoptosis of parasite (Gonzalez et al. Reference Gonzalez, Desponds, Schaff, Mottram and Fasel2007). Since metacaspase are associated with chromosomal separation and survival of the parasite they can directly be used as a drug target(Meslin et al. Reference Meslin, Zalila, Fasel, Picot and Bienvenu2011). So the molecules that can target on the biosynthetic machinery of metacaspase might be used as the effective antileishmanial agent. However, more studies are required to understand the entire role of leishmanial metacaspases.
Enzymes of folate biosynthesis
The key enzyme which is responsible for the production of thymidine in folate metabolism is DHFR (Booth et al. Reference Booth, Selassie, Hansch and Santi1987). Using NADPH as cofactor it reduces dihydrofolate to tetrahydrofolate. As a result, DHFR inhibition stops thymidine biosynthesis and as a consequence, biosynthesis of DNA. Auspiciously DHFR has been crystallized from Trypanosoma cruzi and L. major and structural data between parasite and human enzymes may be achieved so that inhibitors of DHFR can be designed (Senkovich et al. Reference Senkovich, Schormann and Chattopadhyay2009). An enzyme dihydrofolate reductase-thymidylate (DHFR-TS) is also associated with the survival of parasite and without this enzyme parasites are not able to survive in animal hosts (Veras et al. Reference Veras, Brodskyn, Balestieri, Freitas, Ramos, Queiroz, Barral, Beverley and Barral-Netto1999). In addition, an approach using database mining to explore novel inhibitors of DHFR has also been made but it needs more focus to use DHFR as a drug target (Zuccotto et al. Reference Zuccotto, Martin, Laskowski, Thornton and Gilbert1998). Despite these advantages, various resistance mechanisms to DHFR have been uncovered such as overexpression of the enzyme PTR-1 and DHFR-TS (Wang et al. Reference Wang, Leblanc, Chang, Papadopoulou, Bray, Whiteley, Lin and Ouellette1997). The enzyme PTR-1 not only causes the reduction of biopterin to dihydrofolate and tetrahydrofolate but also reduces dihydrofolate to tetrahydrofolate. Therefore DHFR and PTR-1 can be used as more efficient drug targets.
Topoisomerase
DNA topoisomerases are involved in various biological processes like DNA replication, transcription, recombination, and repair and classified as type I and type II. DNA topoisomerases have been used as chemotherapeutic targets against Leishmania (Das et al. Reference Das, Dasgupta, Sengupta and Majumder2004). Camptothecin, a plant alkaloid which is an inhibitor of eukaryotic topoisomerase-I shows some inhibitory properties against T. brucei, T. cruzi and L. donovani (Bodley et al. Reference Bodley, Wani, Wall and Shapiro1995). A pentacyclic triterpenoid, Dihydrobetulinic acid (DHBA) which is a derivative of betulinic acid shows inhibitory properties against both Topoisomerase I and topoisomerase II of L. donovani (Chowdhury et al. Reference Chowdhury, Mandal, Goswami, Ghosh, Mandal, Chakraborty, Ganguly, Tripathi, Mukhopadhyay, Bandyopadhyay and Majumder2003). Three isoflavanoids, smiranicin, lyasperin H and 8-prenylmucronulatol have been found to be observed with anti-leishmanial attribute and this activity has been correlated to inhibition of topoisomerase II and kDNA linearization (Salem and Werbovetz, Reference Salem and Werbovetz2005). Thus these enzymes should be structurally analysed so that specific inhibitors against Leishmania can be designed.
Protein kinases
Cyclin-dependent kinases (CDKs) are reported to play a significant role in cell division cycle, differentiation, transcription, and apoptosis. For their own activation, few CDKs requires active cyclins and for functional activation, few require phosphorylation which could be accomplished at their conserved threonine residues (t-loop) by cdc2 activating kinases (Affranchino et al. Reference Affranchino, Gonzalez and Pays1993). So in Leishmania, these cdc-2 activating kinases which are essential for the progression of the cell cycle can be used as potential drug targets. Two putative CDKs, LmexCRK3 and LmexCRK1 (Hassan et al. Reference Hassan, Fergusson, Grant and Mottram2001) have also been found to be essential to the promastigotes form. CRK3 belongs to indirubin class, is found to be active throughout the life cycle in L. mexicana (Grant et al. Reference Grant, Hassan, Anderson and Mottram1998; Hassan et al. Reference Hassan, Fergusson, Grant and Mottram2001). The chemical inhibitors of CRK3 deteriorate parasitic viability within macrophage, thus CRK3 can be used as a potential drug target. In another study on L. donovani, recently it was found that glycogen synthase kinase (LdGSK3) is also associated with the control of cell cycle and apoptosis based on the indirubin test (Xingi et al. Reference Xingi, Smirlis, Myrianthopoulos, Magiatis, Grant, Meijer, Mikros, Skaltsounis and Soteriadou2009). Thus, LdGSK3 in combination with CRK3 can be used as possible drug targets. Similarly, other cdks can also be diagnosed as potential drug targets.
Mitogen-activated proteins kinases (MAPK)
Mitogen-activated protein (MAP) kinases play a significant role in signal transduction. Moreover, they act as a regulator of cell differentiation, proliferation and apopotosis. Until now, 15 MAP kinases have been identified in L. mexicana (Wiese, Reference Wiese2007) and the most studied are LmxMPK , LmxMPK9 and LmxMKK (Bengs et al. Reference Bengs, Scholz, Kuhn and Wiese2005). These MAP kinases can further be evaluated as a potential drug target.
Proteinases
Proteinases are of four main types: serine, aspartate, cysteine and metalloenzyme. In case of parasitic protozoans most identified and characterized are cysteine proteinases (CPs). Because of structurally different from mammalian homologue and their role as a putative virulence factor in parasite–host cell interaction CPs have become of great interest as a potential drug target. More than 65 CPs have been observed from the genome sequence analysis of L. major and are further categorized into various types viz. CPA, CPB and CPC. Furthur gene replacement studies of L. mexicana revealed the role of CPA, CPB and CPC in host–parasite interaction. In a study when multicopy CPB gene array (Dcpb) was replaced by L. mexicana, BALB/c mice was observed with poor lesion growth with reduced virulence (Alexander et al. Reference Alexander, Coombs and Mottram1998). Some highly potent inhibitors (Semicarbazones, Thiosemicarbazones and Triazine Nitriles) of L. mexicana cysteine protease CPB have been reported that may also have useful efficacy against other important cysteine proteases (Schröder et al. Reference Schröder, Noack, Marhöfer, Mottram, Coombs and Selzer2013).
Methionine aminopeptidase 2 (MetAP-2)
MetAP-2 is a cellular metalloexopeptidase, participates in the late hydrolysis of the initiator methionine of protein synthesis (Walker KF, Reference Walker, Arfin and Bradshaw1998). Fumagillin which is an inhibitor of MetAP-2 prevent the replication of L. donavani (Zhang et al. Reference Zhang, Nicholson, Bujnicki, Su, Brendle, Ferdig, Kyle, Milhous and Chiang2002).
Microtubule-associated protein (MAP2)
In past, parasitic microtubules have been identified as a potential target but due to the high toxicity of antimicrotubule drugs, its further use in regular practice was restricted. MAP2, an ethyl 3-(chloroacetamido)-benzoate that is effective on diverse species of Leishmania, alters the organization of microtubule in the parasite. Its in vivo activity was observed in an L. major mice model by the important reduction of the parasite load in the liver, spleen and lymph node (Hiam et al. Reference Hiam, Sebastien, George, Arlette, Kalil and Le Pape2006).
Enzymes of sterol biosynthesis
The major growing sterols in Leishmania are ergosterol and stigmasterol, which is different from mammalian complimentary cholesterol. This can be used as an effective drug target. Ergosterol has two main functions: (1) it is a construction unit of the cell membrane and (2) it might play a significant role in the hormonal activity. Because of sterol based mechanism, AmB reveals its effectiveness against Leishmaniasis but due to AmB resistance for Leishmania, it orders investigators to search an alternative drug of AmB. Azasterol, a class of s-adenosyl-L-methionine performs antileishmanial activity by inhibiting 24-methyltransferase (the vital enzyme in ergosterol biosynthesis) (Lorente et al. Reference Lorente, Jimenez, Gros, Yardley, de Luca-Fradley, Croft, A Urbina, Ruiz-Perez, Pacanowska and Gilbert2005). Other compounds such as azole and triazole inhibit 14α-methylsterol 14-demethylase. Thus are effective against Leishmania. During the development of the novel drug, it must be in consideration that when the sterol profile of Leishmania get alters it metabolizes host sterol for its survival (Roberts et al. Reference Roberts, Jiang, Gasteier, Frydman, Marton, Heby and Ullman2007).
DEAD BOX RNA helicase
DEAD-box RNA helicases are important in tumorigenesis and viral infection (Ariumi, Reference Ariumi2014) so it represents a potent novel therapeutic target (Xu et al. Reference Xu, Peng, Wang, Fang and Karbowski2011). In the case of Leishmania, ATP-dependent DEAD-box RNA helicase plays important role in RNA metabolism and cellular signalling. Recently it has been reported that a 67 kDa DEAD-box ATP-dependent RNA helicase preventing antisense rRNA cleavage, and protecting rRNA from degradation and Leishmania from cell death (Padmanabhan et al. Reference Padmanabhan, Samant, Cloutier, Simard and Papadopoulou2012). Further Leishmania DDX3 homologue of DEAD-box RNA helicase plays a central role in mitochondrial protein quality control under normal growth conditions and particularly upon stress by preventing ROS-mediated damage and polyubiquitinated protein accumulation in the mitochondrion (Padmanabhan et al. Reference Padmanabhan, Zghidi-Abouzid, Samant, Dumas, Aguiar, Estaquier and Papadopoulou2016). Our unpublished QSAR study also suggested many inhibitors for DEAD-box RNA helicase.
Casein kinase 1 isoform 2 (CK1.2)
Casine kinase is a member of the highly conserved Ser/Thr protein kinase family which play important role in signal transduction (Knippschild et al. Reference Knippschild, Gocht, Wolff, Huber, Löhler and Stöter2005). CK1 contains six isoforms in Leishmania. LmjF35.1010 (LmCK1.2), the major isoform, has been validated pharmacologically as a drug target because CK1 shown to be essential for intracellular parasite survival and infectivity (Durieu et al. Reference Durieu, Prina, Leclercq, Oumata, Gaboriaud-Kolar, Vougogiannopoulou, Aulner, Defontaine, No and Ruchaud2016). Two lead compounds have also been identified and selected as LmCK1.2 inhibitors based on their specificity, antileishmanial activity, the absence of cytotoxicity (Durieu et al. Reference Durieu, Prina, Leclercq, Oumata, Gaboriaud-Kolar, Vougogiannopoulou, Aulner, Defontaine, No and Ruchaud2016). There is a further need to understand their mode of action and these compounds could also be used as pharmacological tools to study parasite-specific signal transduction.
L-asparaginase
L-asparaginases enzyme belongs to amidohydrolases family that catalyse the conversion of L-asparagine into L-aspartic acid and ammonia (Vimal and Kumar, Reference Vimal and Kumar2017). The presence of genes coding for putative l-asparaginase enzymes in the L. donovani genome has given a hin towards the specific role of this enzyme in the survival of the parasite. Singh et al. (Reference Singh, Khan, Singh Yadav, Srivastava, Sinha, Ashish Das and Kundu2017) have advocated that L-asparaginases could be a potential drug target against the pathogen Leishmania.
Challenges in antileishmania drug development
The current pace of antileishmanial drug development is very slow and it should be kept up with the clinical needs, particularly as resistance to current agents is being reported frequently and drug resistance mechanisms should be explored. The further slow pace of new antileishmanial drug development is due to: (1) different clinical manifestations, (2) a deficient market, (3) failure of public-health policy, (4) lack of public–private partnerships (PPPs) and (5) lack of technology transfer.
Clinical problem
Leishmaniasis is caused by 21 of 30 species of Leishmania that infect mammals (https://www.cdc.gov/dpdx/leishmaniasis/index.html). These include VL caused by the L. donovani complex; MCL is caused by parasite belongs to the braziliensis complex, such as L. braziliensis, L. panamensis, L. peruviana; and CL is caused by L. major in the Old World but in America this clinical form could be caused by parasite belongs to the subgenus Leishmania and Viannia. The different species are morphologically indistinguishable, but they can be differentiated by isoenzyme analysis, DNA sequence analysis or monoclonal antibodies (Hijjawi et al. Reference Hijjawi, Kanani, Rasheed, Atoum, Abdel-Dayem and Irhimeh2016). Among CL, MCL and VL, VL is a fatal form in which the parasites have migrated to the vital organs (liver, bone marrow and spleen) of the body (Kumar et al. Reference Kumar, Misra, Sisodia, Shasany, Sundar and Dube2015). Up to 90% of the world's cases of VL are found in Bangladesh, Brazil, India, Nepal and Sudan. Wide and vast clinical manifestation of this disease is one major challenge in antileishmanial drug development (Pace, Reference Pace2014).
Deficient market
Food and Drug Administration (FDA), USA approved thousands of new chemical entities from 1995 to 2016, but only a few were for infectious diseases. Among these anti-infectious agents, Miltefosine was approved as the only antileishmanial agent in 2014 in last 21 years (http://www.centerwatch.com/drug-information/fda-approved-drugs/year/2016). There is a more than the 10-fold greater chance of a drug being brought to market for cancer, neurological disorders than for infectious disease. This number is negligible and matter of sorrow for the neglected disease leishmaniasis. Because the pharmaceutical industry argues that drug development is risky and too costly to invest in the low-return area such as leishmaniasis. This a most prominent reason for a limited number of new antileishmanial drugs marketed. The drugs for the neglected diseases working group is currently exploring the feasibility of not-for-profit initiative internationally that would focus on drug development projects for the most the neglected diseases like leishmaniasis. Such an initiative would remove the process of life-saving drugs development from a market-driven logic.
Lack of strong public-health policy
Public-health policy has an intense impact on health status. Good policies are not in use in developing and underdeveloped countries. Most of the developing countries spent <$20 on all health programme per year per person (Sachs, Reference Sachs2002). In these circumstances, the market is too small to attract investors in the development of the drug for the diseases that mainly affect developing countries. The chronic neglected disease crisis calls for a substantial health policy and long-term response worldwide that and will definitely create a paradigm shift in health, research and development. The evidence sheds light that the public-health policy issues are limited in developing and underdeveloped countries. There should be an international pharmaceutical policy for all neglected diseases and a PPP sector come up and promote not-for-profit research and development capacity.
Public–private partnerships
Measures should be taken to overcome the above-discussed constraint and PPP could be a solution. It attempts to fill the gaps in the health needs of developing countries through the establishment of public-private networks and collaboration. The public sector includes aid agencies, international organizations, academia and government bodies and the private sector includes pharmaceutical industries, foundations, philanthropic institutions, and charities. Recently, some PPPs have started drug development activities in the pharmaceuticals sector, as a new paradigm. The PPP concept has recently evolved into product-based and more structured collaborations. The tropical disease research (TDR), WHO and the Walter Reed Army Institute of Research (WRAIR), Maryland, USA is a good example of public institutions working in partnership with the private sector on drug development programmes. The antimalarial drug mefloquine was discovered by the WRAIR and later developed jointly with industry and TDR (Gelband et al. Reference Gelband, Panosian and Arrow2004). However, company engagement is limited and doing initiative for AIDS, Malaria and Tuberculosis, because these diseases rank higher in the public-health priorities of developed countries than other neglected diseases and represent a potential market for the industry. For the neglected diseases such as leishmaniasis, PPP engagement and effort should be more and more.
Technology transfer
Capacity building and technology transfer have been promoted for many years by TDR, United Nations Development Programme, and Drugs for Neglected Diseases Initiatives (DNDI). FDA, NIH and several US university technology transfer offices have compiled a list of their technologies that relate to these specific neglected diseases (Allarakhia, Reference Allarakhia2013). This list is expected to function as an additional tool to facilitate the transfer of such technologies to potential licensee institutions worldwide for the development of effective medicine against neglected disease. Adopting approaches to transfer technologies offers the broadest benefit to populations in need. Practical issues confronted by technology transfer would be helpful in the development of products in neglected disease areas.
Conclusion
Despite impressive advances in science and technology we have failed to allocate sufficient resources to fight the diseases leishmaniasis. There are a deficiency and sluggishness of research and development into neglected diseases to control the re-emergence, develop more effective drugs, and overcome resistance for leishmaniasis to shorten the treatment. Only a few drugs are available for the treatment of leishmaniasis and the appearance of drug resistance is further complicating the control of this disease. A wide range of biological assays of the possible compounds should be performed with several strains and different parasite forms, and active compound should be reached to clinical trials immediately. To meet these challenges, a firm commitment by the pharmaceutical industry partnering with academics, novel chemical libraries and combining innovative screening strategies is required to achieve victory. This suggests that there is a need to come up with newer hit/leads which may help furthermore to combat with leishmaniasis and slow down the potential emergence of resistant Leishmania parasites.
Acknowledgements
Authors are thankful to Kumaun University SSJ campus, Almora (Uttarakhand), India and National Institute of Technology (NIT), Raipur (CG), India for providing a facility for this work.
Financial support
This study was financially supported by the Science and Engineering Research Board (SERB, India) young scientist grant YSS/2014/000773 awarded to M.S.. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors have declared no conflict of interest.




 Open access
Open access