
Volume 81 - Issue 5 - October 2017
Contents
Research Article
Leószilárdite, the first Na,Mg-containing uranyl carbonate from the Markey Mine, San Juan County, Utah, USA
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1039-1050
-
- Article
- Export citation
Crystal chemistry of zinc incorporation in strunzite-group minerals containing zeolitic water
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1051-1062
-
- Article
- Export citation
First crystal-structure determination of natural lansfordite, MgCO3·5H2O
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1063-1071
-
- Article
- Export citation
Dzierżanowskite, CaCu2S2 – a new natural thiocuprate from Jabel Harmun, Judean Desert, Palestine Autonomy, Israel
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1073-1085
-
- Article
- Export citation
-
Dzierżanowskite,
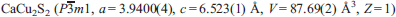 , a thiocuprate, was found in larnite pseudoconglomerate rocks of the Hatrurim Complex at Jabel Harmun, Palestinian Autonomy, Israel. Dzierżanowskite occurs in larnite pebbles, which are embedded in a low-temperature mineral matrix. Associated minerals are larnite, brownmillerite, fluorellestadite, ye'elimite, gehlenite, periclase, ternesite, nabimusaite, vorlanite, vapnikite, fluormayenite, fluorkyuygenite, oldhamite, jasmundite, covellite, chalcocite and pyrrhotite. Electron microprobe analyses yield an average composition of Cu 55.25, Fe 0.13, S 27.46 and Ca 16.99, total 99.83 wt.%. The empirical formula of dzierżanowskite, based on 5 atoms, is Ca0.98Cu2.02Fe0.01S1.99. Dzierżanowskite forms grains up to 15 μm in size or rims on oldhamite and laminar intergrowths with chalcocite and covellite. Dzierżanowskite is dark orange, has a cream streak and a submetallic lustre. In reflected light it is grey, with a cream tint and characteristic yellow-orange internal reflections. The calculated density of dzierżanowskite is 4.391 g cm -3. Three bands at 300, 103 and 86 cm -1 are observed in the Raman spectrum. The strongest lines of the calculated powder diffraction pattern are [d, Å (I) hkl]: 2.358(100) 102, 1.970(93)110, 3.023(78) 011, 6.523(36) 001, 3.412 (28) 100, 1.834(28) 103. Dzierżanowskite was also found in unusual jasmundite rocks, forming small ‘paleofumaroles’ within areas of low-temperature hydrothermal rocks bearing larnite pseudoconglomerates at Jabel Harmun. Dzierżanowskite is a superimposed phase of the high-temperature alteration of pyrometamorphic rocks subjected to by-products (melts/fluids and gases) of pyrometamorphism originating in the deeper levels of combustion.
, a thiocuprate, was found in larnite pseudoconglomerate rocks of the Hatrurim Complex at Jabel Harmun, Palestinian Autonomy, Israel. Dzierżanowskite occurs in larnite pebbles, which are embedded in a low-temperature mineral matrix. Associated minerals are larnite, brownmillerite, fluorellestadite, ye'elimite, gehlenite, periclase, ternesite, nabimusaite, vorlanite, vapnikite, fluormayenite, fluorkyuygenite, oldhamite, jasmundite, covellite, chalcocite and pyrrhotite. Electron microprobe analyses yield an average composition of Cu 55.25, Fe 0.13, S 27.46 and Ca 16.99, total 99.83 wt.%. The empirical formula of dzierżanowskite, based on 5 atoms, is Ca0.98Cu2.02Fe0.01S1.99. Dzierżanowskite forms grains up to 15 μm in size or rims on oldhamite and laminar intergrowths with chalcocite and covellite. Dzierżanowskite is dark orange, has a cream streak and a submetallic lustre. In reflected light it is grey, with a cream tint and characteristic yellow-orange internal reflections. The calculated density of dzierżanowskite is 4.391 g cm -3. Three bands at 300, 103 and 86 cm -1 are observed in the Raman spectrum. The strongest lines of the calculated powder diffraction pattern are [d, Å (I) hkl]: 2.358(100) 102, 1.970(93)110, 3.023(78) 011, 6.523(36) 001, 3.412 (28) 100, 1.834(28) 103. Dzierżanowskite was also found in unusual jasmundite rocks, forming small ‘paleofumaroles’ within areas of low-temperature hydrothermal rocks bearing larnite pseudoconglomerates at Jabel Harmun. Dzierżanowskite is a superimposed phase of the high-temperature alteration of pyrometamorphic rocks subjected to by-products (melts/fluids and gases) of pyrometamorphism originating in the deeper levels of combustion.
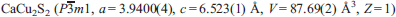 , a thiocuprate, was found in larnite pseudoconglomerate rocks of the Hatrurim Complex at Jabel Harmun, Palestinian Autonomy, Israel. Dzierżanowskite occurs in larnite pebbles, which are embedded in a low-temperature mineral matrix. Associated minerals are larnite, brownmillerite, fluorellestadite, ye'elimite, gehlenite, periclase, ternesite, nabimusaite, vorlanite, vapnikite, fluormayenite, fluorkyuygenite, oldhamite, jasmundite, covellite, chalcocite and pyrrhotite. Electron microprobe analyses yield an average composition of Cu 55.25, Fe 0.13, S 27.46 and Ca 16.99, total 99.83 wt.%. The empirical formula of dzierżanowskite, based on 5 atoms, is Ca
, a thiocuprate, was found in larnite pseudoconglomerate rocks of the Hatrurim Complex at Jabel Harmun, Palestinian Autonomy, Israel. Dzierżanowskite occurs in larnite pebbles, which are embedded in a low-temperature mineral matrix. Associated minerals are larnite, brownmillerite, fluorellestadite, ye'elimite, gehlenite, periclase, ternesite, nabimusaite, vorlanite, vapnikite, fluormayenite, fluorkyuygenite, oldhamite, jasmundite, covellite, chalcocite and pyrrhotite. Electron microprobe analyses yield an average composition of Cu 55.25, Fe 0.13, S 27.46 and Ca 16.99, total 99.83 wt.%. The empirical formula of dzierżanowskite, based on 5 atoms, is CaComplex exsolution microstructures in ilmenite–pyrophanite from the Garnet Codera dyke pegmatite (Central Italian Alps): an electron microscopy investigation
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1087-1104
-
- Article
- Export citation
-
Ilmenite–pyrophanite crystals from a garnet pegmatite dyke from the Upper Codera Valley (Sondrio, Italian Alps) showing exsolutions of titanohematite and columbite-tantalite were investigated by scanning and transmission electron microscopy. The titanohematite precipitates share the same crystallographic orientation of the ilmenite-pyrophanite host, are bean-shaped when observed on sections inclined to the pinacoidal section, and are elongated when observed on sections closer to the prism section, possibly because of their discoidal shape parallel to (001). The columbite-tantalite precipitates form a hexagonal network of needles elongated along ⟨110⟩ of the ilmenite–pyrophanite and titanohematite host. The following crystallographic relationship was established: [100]Col//[001]Ilm; [001]Col//[110]Ilm;
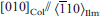 , which can be explained in terms of preservation of the oxygen close packing between the ilmenite and columbite structures. The interfaces between any two of the three different phases are coherent but show lattice strain contrast and sometimes dislocations because of their different unit-cell dimensions. On the basis of textural observations, titanohematite is supposed to exsolve first, followed by columbite-tantalite at temperatures below 500°C. The addition of MnO to the Fe2O3–FeTiO3 system is supposed to considerably influence the topology of the related T-X phase diagram and the solubility of Nb2O5 and Ta2O5 in this system.
, which can be explained in terms of preservation of the oxygen close packing between the ilmenite and columbite structures. The interfaces between any two of the three different phases are coherent but show lattice strain contrast and sometimes dislocations because of their different unit-cell dimensions. On the basis of textural observations, titanohematite is supposed to exsolve first, followed by columbite-tantalite at temperatures below 500°C. The addition of MnO to the Fe2O3–FeTiO3 system is supposed to considerably influence the topology of the related T-X phase diagram and the solubility of Nb2O5 and Ta2O5 in this system.
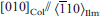 , which can be explained in terms of preservation of the oxygen close packing between the ilmenite and columbite structures. The interfaces between any two of the three different phases are coherent but show lattice strain contrast and sometimes dislocations because of their different unit-cell dimensions. On the basis of textural observations, titanohematite is supposed to exsolve first, followed by columbite-tantalite at temperatures below 500°C. The addition of MnO to the Fe
, which can be explained in terms of preservation of the oxygen close packing between the ilmenite and columbite structures. The interfaces between any two of the three different phases are coherent but show lattice strain contrast and sometimes dislocations because of their different unit-cell dimensions. On the basis of textural observations, titanohematite is supposed to exsolve first, followed by columbite-tantalite at temperatures below 500°C. The addition of MnO to the FeCentennialite, CaCu3(OH)6Cl2.nH2O, n ≈ 0.7, a new kapellasite-like species, and a reassessment of calumetite
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1105-1124
-
- Article
- Export citation
-
The new mineral centennialite (IMA 2013-110), CaCu3(OH)6Cl2·nH2O, was identified from three cotype specimens originating from the Centennial Mine, Houghton County, Michigan, USA, where it occurs as a secondary product, after acid water action upon supergene Cu mineralization in association with, and essentially indivisible from, other copper-containing minerals such as calumetite and atacamite family minerals. It forms as pale to azure blue encrustations, often taking a botryoidal form. Centennialite is trigonal,
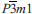 , a = 6.6606(9) Å, c = 5.8004(8) Å, V= 222.85(6) Å3, Z= 1. The strongest powder X-ray diffraction lines are dobs/Å [I%] (hkl), 5.799 [100] (001), 2.583 [75] (201), 2.886 [51] (111), 1.665 [20] (220), 1.605 [17] (023), 1.600 [15] (221), 1.444 [11] (222). The X-ray refined structure forms a kagome net of planar coordinated CuO4 units with Jahn-Teller coordinated Cl apices to form octahedra that edge-share to in-plane adjacent and flattened CaO6 octahedra, which are centred about the lattice origin. All oxygen sites are protonated and shared between one Ca-octahedron and one CuO4 planar unit. Three protonated sites are linked, by hydrogen-bonding to Cl sites, which sit on the triad axis. Each lattice has one Cl above and one below the Ca-Cu polyhedral plane. Consequently, the layers are stacked, along ⟨001⟩, with two Cl sites between layers. In addition to this kapellasite-like topology, an extra c/2 site is identified as being variably water-hosting and extends the coordination of the Ca-site to 8-fold, akin to the body-diagonal Pb-Cu sheet in murdochite. Centennialite conforms to the description of the ‘Unidentified Cu-Ca-Cl Mineral’ noted in Heinrich's Mineralogy of Michigan and is almost certainly identical to the supposed hexagonal basic calcium-copper hydroxychloride monohydrate of Erdös et al. (1981). We comment upon relationships between calumetite and centennialite and propose a substructure model for a synthetic calumetite-like phase that is related directly to centennialite.
, a = 6.6606(9) Å, c = 5.8004(8) Å, V= 222.85(6) Å3, Z= 1. The strongest powder X-ray diffraction lines are dobs/Å [I%] (hkl), 5.799 [100] (001), 2.583 [75] (201), 2.886 [51] (111), 1.665 [20] (220), 1.605 [17] (023), 1.600 [15] (221), 1.444 [11] (222). The X-ray refined structure forms a kagome net of planar coordinated CuO4 units with Jahn-Teller coordinated Cl apices to form octahedra that edge-share to in-plane adjacent and flattened CaO6 octahedra, which are centred about the lattice origin. All oxygen sites are protonated and shared between one Ca-octahedron and one CuO4 planar unit. Three protonated sites are linked, by hydrogen-bonding to Cl sites, which sit on the triad axis. Each lattice has one Cl above and one below the Ca-Cu polyhedral plane. Consequently, the layers are stacked, along ⟨001⟩, with two Cl sites between layers. In addition to this kapellasite-like topology, an extra c/2 site is identified as being variably water-hosting and extends the coordination of the Ca-site to 8-fold, akin to the body-diagonal Pb-Cu sheet in murdochite. Centennialite conforms to the description of the ‘Unidentified Cu-Ca-Cl Mineral’ noted in Heinrich's Mineralogy of Michigan and is almost certainly identical to the supposed hexagonal basic calcium-copper hydroxychloride monohydrate of Erdös et al. (1981). We comment upon relationships between calumetite and centennialite and propose a substructure model for a synthetic calumetite-like phase that is related directly to centennialite.
 ,
, The discreditation of girdite
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1125-1128
-
- Article
- Export citation
Synthesis and crystal structure of C2/c Ca(Co,Mg)Si2O6 pyroxenes: effect of the cation substitution on cell volume
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1129-1139
-
- Article
- Export citation
Currierite, Na4Ca3MgAl4(AsO3OH)12·9H2O, a new acid arsenate with ferrinatrite-like heteropolyhedral chains from the Torrecillas mine, Iquique Province, Chile
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1141-1149
-
- Article
- Export citation
Omariniite, Cu8Fe2ZnGe2S12, the germanium analogue of stannoidite, a new mineral species from Capillitas, Argentina
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1151-1159
-
- Article
- Export citation
Correction
Erratum to Gibson (2017) On the nature and origin of garnet in highly-refractory Archean lithospheric mantle: constraints from garnet exsolved in Kaapvaal craton orthopyroxenes (Mineralogical Magazine, 81, 781–809).
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1161-1163
-
- Article
-
- You have access
- Export citation
Research Article
Fluorophlogopite-bearing and carbonate metamorphosed xenoliths from theCampanian Ignimbrite (Fiano, southern Italy): crystal chemical, geochemical and volcanological insights
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1165-1189
-
- Article
- Export citation
Crystal structure determination of karibibite, an Fe3+ arsenite, using electron diffraction tomography
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1191-1202
-
- Article
- Export citation
Three-dimensional study by synchrotron radiation computed tomography of melt distribution in samples doped to enhance contrast
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1203-1222
-
- Article
- Export citation
A re-examination of water in agate and its bearing on the agate genesis enigma
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1223-1244
-
- Article
- Export citation
Dissolution kinetics of hydrated calcium aluminates (AFm-Cl) as a function of pH and at room temperature
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1245-1259
-
- Article
-
- You have access
- Open access
- Export citation
Cancrinite–vishnevite solid solution from Cinder Lake (Manitoba, Canada): crystal chemistry and implications for alkaline igneous rocks
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1261-1277
-
- Article
- Export citation
IMA Commission on New Minerals, Nomenclature and Classification (CNMNC) Newsletter 39
CNMNC Newsletter
New minerals and nomenclature modifications approved in 2017
-
- Published online by Cambridge University Press:
- 02 January 2018, pp. 1279-1286
-
- Article
-
- You have access
- Export citation