



Introduction
Monoclonal gammopathies or paraproteinemias are a heterogeneous spectrum of clonal plasma cell disorders ranging from asymptomatic monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) to symptomatic multiple myeloma (MM), POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes), solitary plasmacytoma (SP) and primary systemic amyloidosis (AL). They are characterized by the secretion of a monoclonal protein which is made up of a heavy chain and a light chain. These disorders are diagnosed as per standard diagnostic criteria laid by the International Myeloma Working Group (IMWG). Reference Rajkumar, Dimopoulos and Palumbo1
Peripheral neuropathy is a well-recognized manifestation of paraproteinemia and is termed paraproteinemic neuropathy (PPN). Studies have shown that paraproteins are present in 3% to 4% of the population older than the age of 50 years, more than 5% of the population older than the age of 70 years, and can be detected in approximately 3% to 5% of patients with chronic peripheral neuropathy. Reference Kelly, Kyle, O’Brien and Dyck2,Reference Kyle, Therneau and Rajkumar3 Approximately half of them are causally related, and the remaining are likely to be coincidental. Peripheral nerve involvement is not considered as an end-organ involvement in plasma cell disorders, and hence there is no consensus on whether they should be treated with anti-myeloma therapy. Therefore, treatment strategies for the various subtypes of PPN are also different. The major role of the neurologist is in the evaluation and treatment of neuropathies associated with MGUS. The neurologist also has a key role in the identification of the underlying systemic hematologic disorders when neuropathy is the presenting feature.
There is a lack of systematic studies about the clinical spectrum and outcomes of PPN. Hence, this retrospective analysis was undertaken to study the clinical, biochemical, and electrophysiological profile, along with outcomes PPN. The utility of nerve conduction studies (NCSs) to differentiate between the demyelinating subtypes was also explored.
Methods
The study was approved by the Institutional Review Board. We included all patients aged more than 16 years, presenting to the inpatient neurology services of Christian Medical College Vellore, a quaternary referral center in South India, between January 2010 and December 2019 with clinical features of a chronic peripheral neuropathy and on further evaluation were found to have either a monoclonal protein or a plasmacytoma. Those with a prior diagnosis of a plasma cell disorder and subsequently developing neuropathy were excluded. All patients were evaluated for alternate etiologies of neuropathy, and those patients with other identified etiologies were excluded. Patient demographics, laboratory profile, treatment, and follow-up details were retrieved from the hospital electronic medical records.
All the patients had NCS using conventional techniques. Parameters measured included distal motor latency (DML), motor conduction velocities (MCVs), compound muscle action potentials (CMAPs), F-wave latencies, and sensory nerve action potentials. The presence of a conduction block, defined as a ≥50% reduction in CMAP amplitude between proximal and distal when the distal CMAP ≥1 mV, was also recorded. The neuropathy was defined as demyelinating if it satisfied the electrophysiological criteria for chronic inflammatory demyelinating polyneuropathy (CIDP). 4 Terminal latency index (TLI) for each motor nerve was calculated as distal conduction distance (mm)/(MCV [m/s] × DML [ms]). TLI is used to evaluate for slowing involving the distal nerve segment and adjusts the DML for the terminal distance and proximal nerve MCV. Reference Mauermann, Sorenson and Dispenzieri5
The ancillary tests included monoclonal protein testing by serum protein electrophoresis (SPEP), serum-free light chain assay (SFLC), urine Bence Jones proteins (BJP) estimation, and serum immunofixation electrophoresis (SIFE); CSF analysis; comprehensive skeletal survey; and bone marrow evaluation. Based on these investigations, patients were classified into subtypes as per the IMWG criteria. Reference Rajkumar, Dimopoulos and Palumbo1
Patients with MM, POEMS syndrome, and AL were referred to the hematologist and were managed as per standard guidelines. Reference Rajkumar6–Reference Gertz8 Among the other subtypes, patients with demyelinating neuropathy were treated with immunotherapy as per guidelines, 9 while those with axonal neuropathy were given a trial of immunotherapy if the neuropathy was severe and progressive. Patients with SP also received radiotherapy for the plasmacytoma.
The severity of neuropathy at the initial assessment and the last follow-up was documented using the Hughes disability scale (HDS). Reference Hughes, Newsom-Davis, Perkin and Pierce10 The outcomes analyzed were the therapeutic response of neuropathy to treatment and residual neurologic disability. The response of neuropathy to treatment at the time of the last follow-up was classified as improving (increase in HDS score by at least 1), stable (same HDS score), or worsening (decrease in HDS score). Residual neurologic disability was graded into minor (HDS 1, 2) and severe (HDS 3, 4). Only patients with a minimum follow-up of 6 months were included for outcome analysis. Hematological response to anti-myeloma therapy was classified as complete response, very good partial response, partial response, no response, and relapse as per the IMWG Uniform Response Criteria for Multiple Myeloma. Reference Durie, Harousseau and Miguel11
Statistical analysis was performed using IBM Corp. Released 2012, IBM SPSS Statistics for Windows, Version 21.0.Armonk, NY: IBM Corp. Descriptive summaries are presented as frequencies and percentages for categorical variables and mean (standard deviation) or median (minimum–maximum) for continuous variables as appropriate. Comparisons between groups for continuous variables were assessed using ANOVA when parametric tests were required and Kruskal–Wallis test when non-parametric tests were required, as indicated. For categorical variables, Pearson Chi-square test or Fisher’s Exact test was used as indicated. All tests were 2-sided, and a p < 0.05 was considered statistically significant.
Results
Patient Characteristics
A total of 560 patients with chronic peripheral neuropathy were evaluated during the study period, out of which 74 patients (13%) were diagnosed with a PPN. There were 61 males (82.4%) and 13 females (17.6%) with a median age of 51.5 years (17–80).
Neuropathy Phenotype and HDS Score
The median duration of symptoms was 12 months (2–72). At the initial presentation, the median HDS score was 3. The clinical phenotype at presentation was motor-sensory in 56 patients (75.7%), while it was a pure sensory presentation in the remaining 18 (24.3%). Of the 18 patients with a pure sensory neuropathy, 10 had a distal sensory involvement, 4 had a sensory ataxic presentation and the remaining 4 had a pure small fiber involvement. The patients with small fiber neuropathy had refractory symptoms with autonomic dysfunction, a normal NCS, and abnormal sympathetic skin response. Intra-epidermal nerve fiber density and quantitative sensory testing were not available at our center. They were classified as probable small fiber neuropathy as per the diagnostic criteria. Reference Tesfaye, Boulton and Dyck12
Electrophysiological Features
On NCS, 51 patients (68.9%) showed demyelinating features and 19 (25.7%) showed axonal features. In the remaining 4 patients (5.4%), NCS was normal and all of them were presumed to have a small fiber neuropathy. Of the 51 patients with demyelinating features, 32 patients were previously diagnosed elsewhere as idiopathic CIDP. This included 13 with MGUS (2 with IgM MGUS and 11 with Non-IgM MGUS), 10 with POEMS, 8 with SP, and one with MM.
Monoclonal Proteins
Of the 74 patients, we were able to identify a monoclonal protein in 44 patients (59.5%) by SPEP and SFLC. In an additional 24 patients (32.4%), they were identified only by SIFE. Urine BJP was positive only in 8 patients (10.8%). The most common heavy chain class was IgG, present in 43 patients (58.1%), followed by IgA in 12 (16.2%), and IgM in 4 (5.4%). The light chain type was lambda in 43 (58.1%) and kappa in 25 (33.8%). Nine patients (12.1%) had a pure light chain monoclonal protein.
Subtype Classification
Based on the IMWG criteria, the subtypes of PPN in our cohort were MGUS (n = 34, 45.9%); POEMS syndrome (n = 18, 24.3%); SP (n = 13, 17.6%); MM (n = 6, 8.1%); and AL (n = 3, 4.1%). The clinical, electrophysiological, and laboratory characteristics of the subtypes are shown in Table 1 and Table 2.
Table 1: Clinical and electrophysiological characteristics of patients with PPN

NP = neuropathy phenotype; EDx = electrodiagnosis; DM = demyelinating; AX = axonal.
* Includes patients with MM and SMM.
# Four patients with MGUS have a pure small fiber neuropathy with normal NCS.
Table 2: Monoclonal protein and laboratory characteristics of patients with PPN
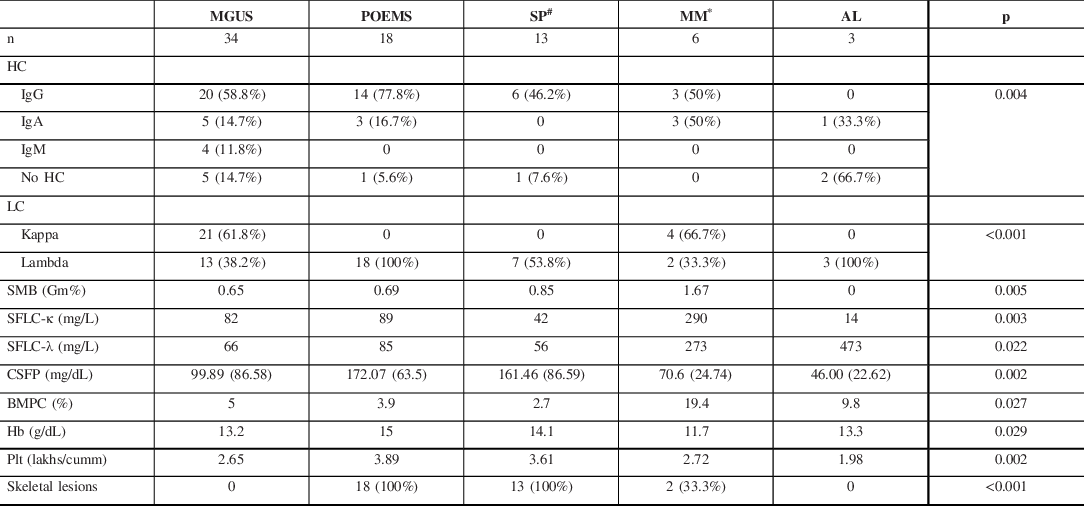
HC = Heavy chain; LC = light chain; SMB = mean M band on SPEP; SFLC = mean serum-free light chain; CSFP = mean CSF protein; BMPC = mean bone marrow plasma cells; Hb = mean hemoglobin; Plt = mean platelet count.
# Six patients with SP did not have detectable monoclonal proteins.
* Includes patients with MM and SMM.
MGUS-Related Neuropathy
Patients with MGUS were heterogeneous with their clinical, biochemical, and electrophysiological features. On NCS, 17 (50%) had demyelinating features, out of which 3 had IgM MGUS and the remaining had non-IgM MGUS. In the remaining 17 patients, 13 had axonal changes and 4 had a normal NCS (pure small fiber neuropathy). Twenty-one patients were treated with immunotherapy, while the remaining 13 received symptomatic treatment. All patients on immunotherapy received corticosteroids as first-line, with 3 receiving combined corticosteroids and IVIG and 2 receiving combined corticosteroids and plasma exchange. Regarding second-line agents, 15 patients received cyclophosphamide and the remaining 6 received rituximab.
POEMS Syndrome
Patients with POEMS syndrome were younger compared to the other subtypes. The monoclonal protein was lambda light chain type in all patients. All of them had a demyelinating neuropathy and presented with severe neuropathic features, with an HDS score of 3 (33.3%) or 4 (66.7%) at presentation. CSF protein levels were elevated to a greater extent as compared to the other subtypes. All 18 patients had multiple sclerotic bone lesions (Figure 1) and skin changes; 14 had organomegaly; 10 had volume overload; 5 had thrombocytosis; 7 had erythrocytosis; and 3 had papilledema. Serum VEGF assay was done in 5 patients and was found to be elevated in 2 of them.
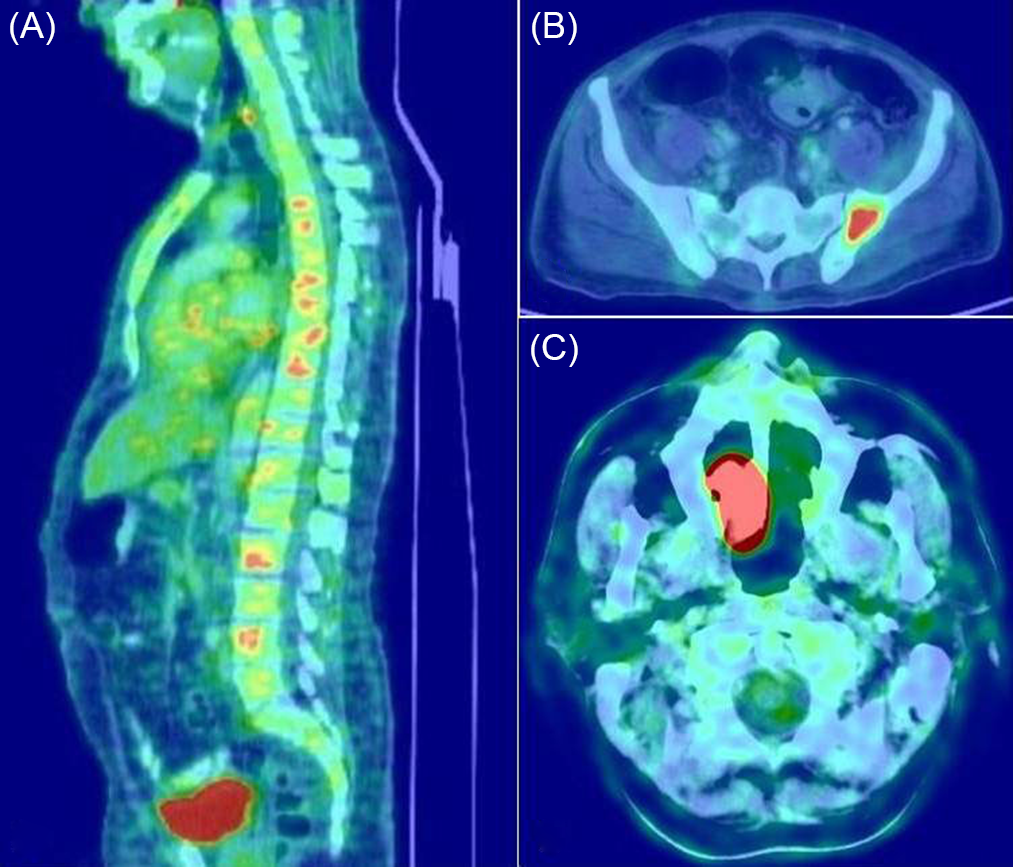
Figure 1: The utility of skeletal survey with FDG-PET/CT in PPN. Multiple sclerotic bone lesions in a patient with POEMS syndrome (A), solitary bone plasmacytoma in the pelvic bone (B), solitary extramedullary plasmacytoma in the nasal cavity (C).
SP-Associated Neuropathy
SP can occur as solitary bone plasmacytoma or as an extramedullary (extraosseous) plasmacytoma. The site of the SP was the vertebral bones in 7 patients; pelvic bones in 2; and the ribs, femur, scapula, and nasal cavity in 1 patient each (Figure 1). Seven patients had a lambda light chain monoclonal protein, whereas, in the remaining 6 patients, a monoclonal protein could not be detected by the laboratory tests. All patients with SP-associated PPN also had a demyelinating neuropathy, and the majority had a severe presentation, with HDS of 3 (30.8%) and 4 (46.2%).
MM-Associated Neuropathy
Six patients were diagnosed with MM, out of which one patient had SMM. The majority presented with severe neuropathic symptoms, with 66.7% having an HDS score of 3. NCS showed axonal changes in 3 and demyelination in the remaining 3 patients.
AL Amyloid Neuropathy
The monoclonal protein was a lambda light chain in all three patients with AL amyloid neuropathy. The presentation was with refractory small fiber neuropathy along with autonomic involvement. All of them had cardiac involvement, and one had features of nephrotic syndrome. Nerve biopsy did not show amyloid in all these patients, and the diagnosis was established by rectal biopsy in 2 patients and lip biopsy in one patient.
Nerve Conduction Studies
The NCS parameters of the paraproteinemic demyelinating neuropathy associated with MGUS (n = 17), POEMS syndrome (n = 18), and SP (n = 13) are shown in Table 3. NCS in patients with POEMS syndrome and SP showed a pattern of mixed demyelinating and axonal features. All patients with POEMS syndrome had unobtainable tibial and peroneal CMAPs, while in SP, peroneal CMAPs were unobtainable in 9 (76.9%) and tibial CMAPs in 12 (92.3%). In POEMS syndrome, the median and ulnar MCVs were slightly slower, TLIs were slightly higher, and conduction blocks were more common when compared to the other demyelinating subtypes.
Table 3: NCS parameters of paraproteinemic demyelinating neuropathy associated with MGUS, POEMS syndrome, and SP
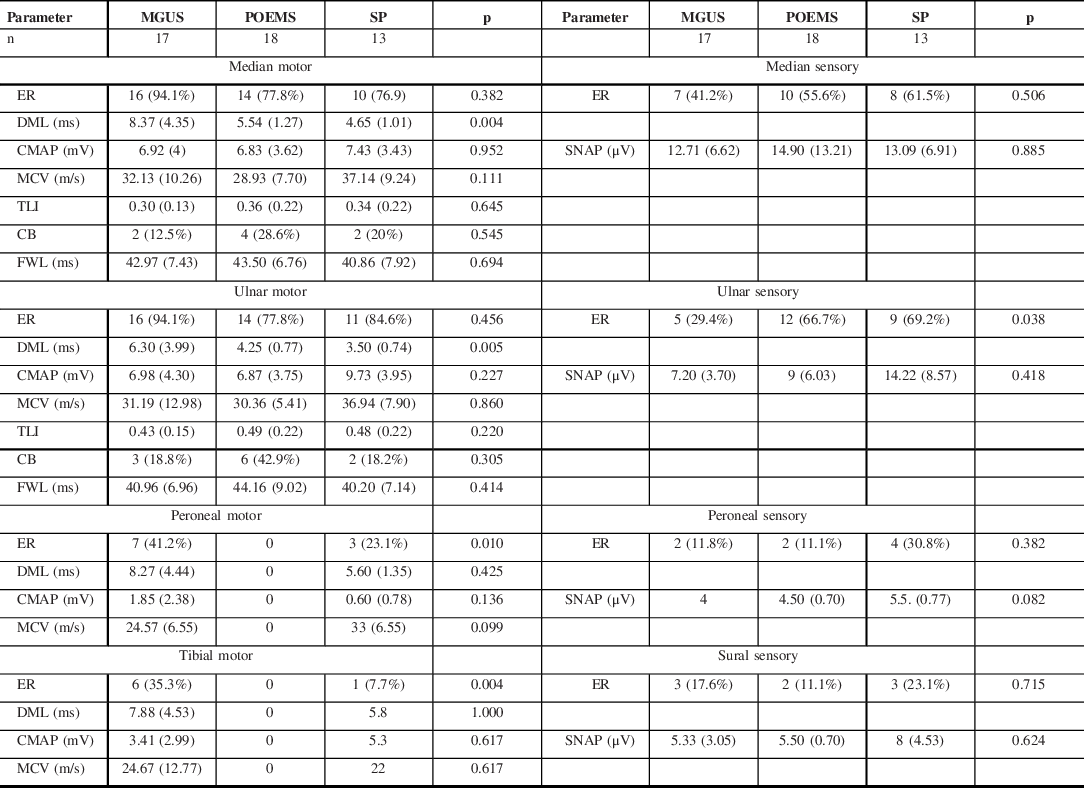
ER = elicitable response; DML = distal motor latency; CMAP = compound muscle action potential; SNAP = sensory nerve action potential; MCV = motor conduction velocity; TLI = terminal latency index; CB = conduction block; FWL = f-wave latency.
The DMLs were more prolonged in the MGUS subtype. A comparison between the median nerve motor NCS features of IgM and non-IgM MGUS subgroups is shown in Table 4. This showed that the IgM subgroup accounted for this finding and they had a greater degree of prolongation of DMLs and lower TLIs in comparison to non-IgM MGUS.
Table 4: Median nerve motor NCS parameters of MGUS-related demyelinating neuropathy according to heavy chain class
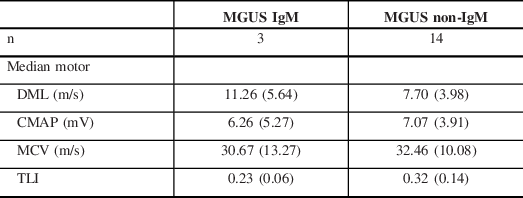
DML = distal motor latency; CMAP = compound muscle action potential; MCV = motor conduction velocity; TLI = terminal latency index.
Disease Course and Outcomes
The anti-myeloma therapy administered to patients with MM, POEMS syndrome, and AL, and the hematological responses are summarized in Table 5. During follow-up, 3 patients evolved into other plasma cell disorders. One patient with MGUS evolved into POEMS syndrome, another evolved into MM, while one with SP developed features of POEMS syndrome. All patients who evolved into POEMS syndrome had lambda light chain type.
Table 5: Anti-Myeloma treatment protocols and responses of MM, POEMS syndrome, and AL amyloidosis subtypes

CTD = Cyclophosphamide, Thalidomide, Dexamethasone; CyBorD = Cyclophosphamide, Bortezomib, Dexamethasone; LD = Lenalidomide, Dexamethasone; Len-LipoDox-Dex = Lenalidomide, Liposomal Doxorubicin, Dexamethasone; ASCT = Autologous stem cell transplant; NR = no response; PR = partial response; VGPR = very good partial response; CR = complete response.
# Evolved from MGUS.
Forty-nine patients had a follow-up of more than 6 months and were included in the outcome analysis. They included MGUS (n = 20, 40.8%), POEMS syndrome (n = 15, 30.6%), SP (n = 9, 18.4%), and MM (n = 5, 10.2%). The median duration of follow-up was 24 months (6–168). The outcomes of therapeutic response and residual neurologic disability are shown in Figure 2. In most patients, the neuropathic symptoms either improved (n = 15, 30.6%) or stabilized (n = 32, 65.3%) with treatment while worsening occurred in two patients (4.1%), one with SP and another with MM. The median HDS score at the last follow-up was also 3. However, many patients had a severe disability (n = 32, 65.3%), and it was highest in patients with POEMS syndrome (n = 13, 86.7%) and lowest in MGUS (n = 8, 40%). The remaining 17 patients (34.7%) had a minor disability. The median duration of illness was 12 months (range 3–60) in patients with severe disability and 12 months (range 2–72) with minor disability (p 0.841).

Figure 2: Outcomes of various subtypes of PPN. Bar diagram showing the HDS score at the initial presentation (i) and last follow-up (l) in various subtypes of PPN (A). Bar diagrams showing the therapeutic response and residual disability in various subtypes of PPN (B, C).
Discussion
This study of a large cohort of hospitalized patients with neuropathy as the presenting symptoms of paraproteinemia has demonstrated the utility of clues from clinical presentation, electrophysiological patterns, biochemical correlations, and a comprehensive skeletal survey in the early diagnosis of different subtypes of PPN. The other striking features that were highlighted include a significant number of patients whose conditions had previously been diagnosed as refractory CIDP, the severity of neurological impairment in certain subtypes, and the relative persistence of neurological disability despite hematological stabilization.
Demographic Details
PPN is a frequently considered etiology during the evaluation of chronic peripheral neuropathy. M proteins have been detected in up to 10% of patients during chronic neuropathy evaluation in tertiary care centers. Reference Kelly, Kyle, O’Brien and Dyck2 In our cohort, 13% of chronic neuropathies were diagnosed with PPN. In contrast to MM and MGUS, POEMS and SP had a relatively younger age of onset as observed in previous studies. Reference Kulkarni, Mahadevan and Taly13 There was also a male preponderance. Reference Gosselin, Kyle and Dyck14
Clinical Phenotype and Electrodiagnosis
Neuropathy can be the presenting feature of paraproteinemia. It is the presenting feature of about 50% of patients with POEMS syndrome, Reference Nasu, Misawa and Sekiguchi15 and 25% of patients with AL amyloid, Reference Rajkumar, Gertz and Kyle16 and can also be seen in about 11 to 13% of untreated MM patients. Reference Chaudhry, Cornblath, Polydefkis, Ferguson and Borrello17 SP is a rare condition and neuropathy as the presenting feature of SP is even rarer. The association between SP and neuropathy had only been described in case reports. Reference Velasco, Bau, Povedano, Petit, Lucas and Bruna18
All patients with POEMS syndrome, SP, and 50% of patients with MGUS and MM had demyelinating features on NCS. They are common CIDP mimics, with over 60% of patients being initially misdiagnosed. Reference Nasu, Misawa and Sekiguchi15 In our cohort, 32 patients were previously diagnosed elsewhere as CIDP and presented with subsequent clinical deterioration.
NCS in patients with POEMS syndrome and SP demonstrates a pattern of mixed demyelinating and axonal features, predominantly in the lower limbs. This reflects the severity of neuropathy and axon loss in these subtypes. Reference Mauermann, Sorenson and Dispenzieri5,Reference Nasu, Misawa and Sekiguchi15
In POEMS syndrome, the slowing of nerve conductions occurs predominantly in the intermediate segments than in the distal nerve segments as evidenced by slower MCVs, higher TLIs (0.35–0.6), and more frequent conduction blocks. Reference Nasu, Misawa and Sekiguchi15 This suggests that demyelination occurs predominantly in the intermediate nerve trunks rather than the distal nerve terminals, where the blood-nerve barrier occurs. The possible mechanism is VEGF mediated diffuse breakdown of the blood-nerve barrier, resulting in homogenous demyelination. Reference Kuwabara and Misawa19
In IgM MGUS-associated demyelinating neuropathy, the clinical presentations of most patients are similar to that of classic CIDP, while some will present with a distal predominant neuropathy termed DADS-M (distal acquired demyelinating symmetric neuropathy–M protein). Reference Katz, Saperstein, Gronseth, Amato and Barohn20 Around 50%–75% of those with DADS-M will have anti-MAG antibodies. Reference Gosselin, Kyle and Dyck14 NCS shows a marked prolongation of the DMLs disproportionate to the proximal segment MCVs and a very low TLI (<0.25), all implying involvement of the distal nerve terminals. Reference Capasso, Torrieri, Di Muzio, De Angelis, Lugaresi and Uncini21
Patients with Non-IgM MGUS can present with a spectrum of phenotypes ranging from the common length-dependent sensorimotor axonal neuropathy to classic CIDP. Reference Gosselin, Kyle and Dyck14 Those with demyelinating electrophysiology usually respond to standard therapies for CIDP despite the presence of the paraprotein, again pointing to the fact that this association may be coincidental. Reference Dyck, Low and Windebank22 In our cohort, there were 14 patients with non-IgM MGUS and a demyelinating neuropathy, in them the association could have been coincidental.
In AL amyloid neuropathy, the somatic large fiber neuropathy is associated with prominent and early small fiber and autonomic involvement as seen in all 3 patients in our cohort. Reference Loavenbruck, Singer and Mauermann23 The phenotype of neuropathy associated with MM is heterogeneous. It can present as a slowly progressive symmetric sensorimotor neuropathy, a primary sensory neuropathy, or rarely as a predominantly motor neuropathy. Reference Kelly, Kyle, Miles, O’Brien and Dyck24 AL amyloid neuropathy is always axonal, while MM usually shows axonal features, rarely, demyelination is seen. Reference Kelly25
Laboratory Tests, Monoclonal Proteins, and Skeletal Survey
The presence of either thrombocytosis or erythrocytosis can be a pointer to POEMS syndrome and is seen in 50% of patients. Reference Dispenzieri, Kyle and Lacy26 In our cohort, 27% had erythrocytosis and 38% had thrombocytosis.
While testing for M protein, SPEP alone can miss many patients with PPN. The IMWG recommends a screening panel that includes SPEP, SIFE, and SFLC. Reference Dispenzieri, Kyle and Merlini27
In POEMS syndrome, a lambda light chain occurs predominantly in 95% and is characteristically IgG or IgA and never IgM. Reference Dispenzieri, Kyle and Lacy26 In our cohort, all the patients had a lambda light chain along with IgG (77.8%) and IgA (16.7%). Thus, the M protein combination of lambda light chain with either IgA or IgG heavy chain is a pointer for POEMS syndrome, and these patients should be thoroughly investigated. This includes a good physical examination, VEGF assay, endocrine profile along with an FDG-PET/CT for bone lesions, organomegaly, and effusions.
The predominant monoclonal light chain type in AL is lambda, seen in about two-thirds of patients. Reference Raheja, Specht and Simmons28 All patients in our cohort with AL amyloid neuropathy also had a lambda light chain. In MM, kappa is the most common light chain and the commonly associated heavy chain is IgG, seen in nearly 50%, followed by IgA. Reference Kyle, Gertz and Witzig29 In our cohort, kappa was the predominant light chain (66.7%) and both IgG and IgA were present equally.
Many studies have shown that the type of M protein in MGUS-associated neuropathy is most commonly IgM, while non-IgM is less common and kappa is more common than lambda. Reference Steiner, Schwärzler, Göbel, Löscher, Wanschitz and Gunsilius30 In our cohort, also kappa chain was more common; however, IgG was the most common heavy chain (58.8%). This may be because IgG is the most common paraprotein and that some of the associations may be coincidental.
Of note, in SP-associated neuropathy, monoclonal proteins are detectable only in about 60%. Reference Soutar, Lucraft and Jackson31 The remaining 40% of patients could remain undiagnosed until the recommended skeletal survey is performed. Moreover, conventional radiography is not sensitive for the detection of extramedullary plasmacytomas. Solitary bone plasmacytoma tends to involve the axial skeleton, most commonly the vertebra, while extramedullary plasmacytoma occurs most commonly in the nasal cavity and nasopharynx. Reference Galieni, Cavo and Pulsoni32
In POEMS syndrome, osteosclerotic lesions occur in approximately 95% of patients and can be easily identified with the use of the recommended skeletal survey. Reference Dispenzieri7 In our cohort, osteosclerotic lesions were present in all patients with POEMS syndrome. The presence of an osteolytic lesion is one of the myeloma defining events for the diagnosis of MM and was present in two patients of our cohort.
Most bone lesions associated with neuropathy are commonly seen in the vertebral bones or pelvis. For the skeletal survey, the IMWG recommends the use of newer imaging techniques like FDG-PET/CT, low-dose whole-body CT, or MRI of the whole-body or spine over conventional radiography. Reference Rajkumar, Dimopoulos and Palumbo1
Disease Course and Outcomes
During follow-up, 2 patients who were initially classified as MGUS and SP evolved into POEMS syndrome and both had lambda light chain type. In the advanced stage of POEMS syndrome, the diagnosis is not difficult; however, it can be challenging in the early stages. This is because of the heterogeneity in clinical presentations, especially presentation with limited features; nonspecific nature of endocrinopathy and skin changes; and misinterpretation of subtle investigation findings. This emphasizes that the presence of demyelinating neuropathy with lambda light chain could be a potential harbinger for later development of POEMS syndrome. One patient with an IgG MGUS evolved to MM.
POEMS syndrome is characterized by a progressive polyneuropathy, with patients developing severe motor weakness leading to greater disability, with some patients becoming wheelchair or bed-bound. Reference Isose, Misawa and Kanai33 Patients may show some improvement in neuropathic features and residual disability with treatment. Reference Karam, Klein and Dispenzieri34 In our cohort, all patients had either improvement or stabilization with treatment; however, 86.7% had a severe disability.
The course of SP-associated neuropathy closely resembles that of POEMS syndrome, with patients developing severe motor weakness. These patients also tend to stabilize or improve with treatment, but the majority have a severe disability.
The course of AL amyloid neuropathy is usually progressive and debilitating. Reference Rajkumar, Gertz and Kyle16 The course of neuropathy in MM can be complicated by treatment-emergent neuropathy occurring in up to 65% of patients receiving chemotherapy, especially with bortezomib and thalidomide. Reference Richardson, Xie and Mitsiades35 However, this aspect was not included in this study.
In IgM MGUS, about 25% to 30% have a moderate residual disability at 10 years after diagnosis. Reference Niermeijer, Fischer, Eurelings, Franssen, Wokke and Notermans36 In our cohort, 8 patients (40%) had a severe disability; the reason maybe because of this being predominantly an inpatient-based cohort.
All the 49 patients included for outcome analysis had a residual disability, with 65.3% having a severe disability. The number of patients with a severe disability is indeed concerning. The possible reasons are an inpatient-based cohort with more severe symptoms as opposed to outpatient setting, delayed diagnosis (median duration of illness of 12 months), and high rates of misdiagnosis, although we could not establish a relationship between the degree of disability and disease duration.
Potential Role of Nerve Biopsy and evolving concept of Monoclonal Gammopathies of ‘Neurological Significance’
Neuropathy as end-organ damage in monoclonal gammopathies has received relatively less attention. Neuropathies at times tend to be severe even though the hematological involvement remains latent. Conventionally, nerve biopsy has been mainly used for the diagnosis of amyloid neuropathy. The yield for the diagnosis of amyloidosis has varied from 30% to 100%. Reference Kyle and Greipp37,Reference Simmons, Blaivas, Aguilera, Feldman, Bromberg and Towfighi38 This has emphasized the need for biopsies of other clinically involved tissues like bone marrow, abdominal fat pad, rectal mucosa, minor salivary gland, or another clinically involved organ for the demonstration of amyloid.
The utility of nerve biopsy in the diagnosis of PPN has extended to include the presence of immunoglobulin complexes by immunohistochemical techniques. Reference Duchesne, Mathis and Corcia39 This could especially be useful in the setting of precursor stages like MGUS and SMM and may also influence earlier initiation of anti-myeloma therapy. The presence of a nerve inflammation pattern as seen in CIDP and the nature of lymphocytic infiltration could also influence treatment algorithms.
Since a majority of patients had a severe disability, an argument could be made in favor of a new classification of these entities as ‘monoclonal gammopathies of neurological significance.’ This may aid in the early diagnosis and the development of individualized targeted treatment regimes, potentially including features from nerve biopsy. It is hypothesized that this may translate into better outcomes and lesser disability. This could be similar to the term monoclonal gammopathy of renal significance which was coined by The International Kidney and Monoclonal Gammopathy Research Group in 2012. This entity comprises hematological conditions that produce a monoclonal immunoglobulin associated with kidney injury in the absence of cast nephropathy. Reference Leung, Bridoux and Hutchison40
The limitations of this study are mainly linked to its retrospective design and an inpatient-based cohort as evidenced by the degree of initial disability. The cohort also had a disproportionately large number of their patients with demyelinating features on NCS and POEMS syndrome subtype. This was probably due to referral bias as this institution is one of the largest referral centers in Southern India. Anti-MAG antibody status and anti-ganglioside antibody status were not available. The rate of progression of MGUS to malignant disease (MM or AL) could not be ascertained as the duration of follow-up was inadequate for the same.
Future Directions
As mentioned, there is a case for earlier definitive treatment of precursor lesions like MGUS and SMM based on the evolution of neuropathy. This could at times obviate the need for immunotherapy with agents like corticosteroids and cyclophosphamide which might, in turn, delay the eventual diagnosis of a hematological malignancy. Further insights into pathophysiology, especially with regard to paraprotein-nerve antigen interactions, could also help in tailoring therapies. This could especially be useful in patients who have a persistent neurological disability even after the achievement of hematological remission in diseases like POEMS and SP. There could be a further role for individualized therapies in patients with PPN in this era of ‘Precision Medicine’.
An algorithmic approach to the diagnosis of PPN is presented in Figure 3. In conclusion, paraproteinemic neuropathies have a heterogeneous spectrum of clinical, biochemical, and electrophysiological features. It should be considered in patients with a refractory CIDP like neuropathy. Following the IMWG recommendation for testing of monoclonal proteins and skeletal survey can help in early diagnosis. NCS can help identify POEMS syndrome and IgM MGUS. In patients with suspected AL amyloid neuropathy, if nerve biopsy is negative, we should strongly consider a biopsy of another clinically involved organ. The majority of patients with PPN tend to stabilize or improve with treatment; however, the residual neurological disability is severe in most patients.
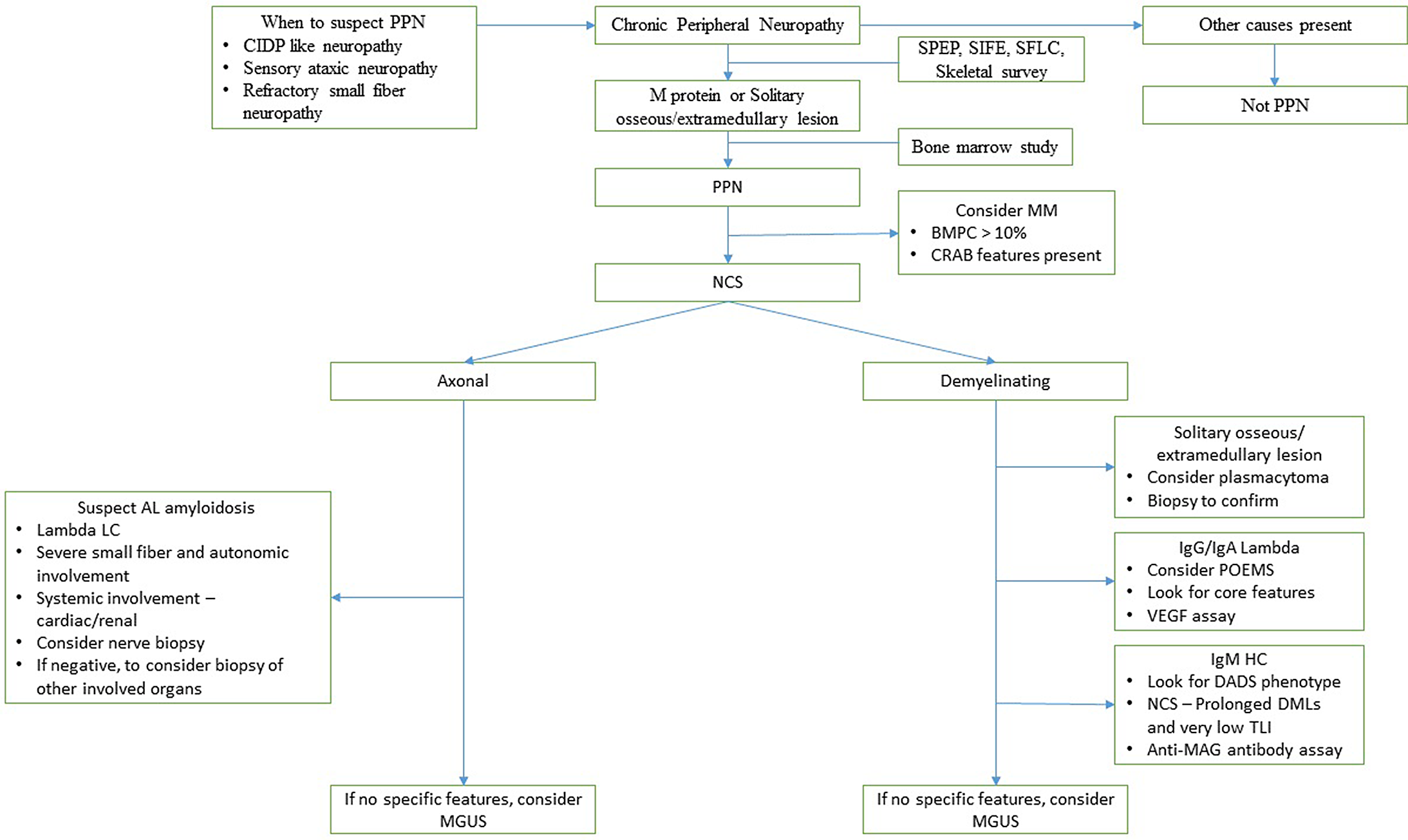
Figure 3: A proposed algorithm for the diagnosis of PPN. (CRAB features are (hypercalcemia, renal failure, anemia, and bone lesions)).
Conflicts of Interest
None.
Statement of Authorship
AMM (Christian Medical College Vellore): design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content. AJD (Christian Medical College Vellore): design and conceptualized study; analyzed the data; drafted the manuscript for intellectual content. AN (Christian Medical College Vellore): revised the manuscript for intellectual content. RNB (Christian Medical College Vellore): revised the manuscript for intellectual content. ATP (Christian Medical College Vellore): revised the manuscript for intellectual content. AS (Christian Medical College Vellore): design and conceptualized study; drafted the manuscript for intellectual content. VM (Christian Medical College Vellore): revised the manuscript for intellectual content. SA (Christian Medical College Vellore): revised the manuscript for intellectual content. BG (Christian Medical College Vellore): revised the manuscript for intellectual content. MA (The Brunei Neuroscience Stroke and Rehabilitation Centre): revised the manuscript for intellectual content.








