


Folate, and its synthetic form, folic acid, is key in one-carbon metabolism, the disruption of which can interfere with DNA synthesis, repair and methylation. Low folate status or, more generally, low dietary methyl status (a combination of folate, methionine, alcohol and other B vitamins) may promote carcinogenesis. The mechanisms by which low folate could increase risk of malignancy include: (1) DNA hypomethylation and inappropriate activation of oncogenesReference Kim1; and/or (2) uracil misincorporation during DNA repair and synthesis, leading to DNA strand breaks, chromosome damage and, eventually, malignant transformationReference Ames2. Higher reported folate intake has been associated with reduced colorectal cancer riskReference Sanjoaquin, Allen, Couto, Roddam and Key3. However, most studies have been conducted in populations where intake is relatively high, and a substantial proportion comes in the form of folic acid, either from supplements or through fortified foods; folic acid is more bioavailable than natural folatesReference Sauberlich, Kretsch, Skala, Johnson and Taylor4.
Efficient one-carbon metabolism also requires riboflavin, vitamin B6, vitamin B12 and methionine. This raises the possibility that these factors, either on their own account, or by acting together with folate, might influence colorectal cancer risk. However, the available evidence is limited and/or inconsistentReference Sharp and Little5.
One-carbon metabolism further requires the optimal activity of multiple enzymes, including 5,10-methylenetetrahydrofolate reductase (MTHFR), which directs the folate pool towards methylation or DNA repair. Several polymorphisms have been reported in the MTHFR gene, but only two have been investigated in relation to colorectal neoplasia – C677T and A1298CReference Sharp and Little5. The 677T variant lowers enzyme activity in vitro Reference Rozen6 and has been associated with decreased plasma folate and vitamin B12 levels and raised homocysteineReference Frosst, Blom and Milos7, Reference Ma, Stampfer and Christensen8. In vitro studies suggest that low folate status may be required for this polymorphism to affect enzyme activity in vivo Reference Guenther, Sheppard, Tran, Rozen, Matthews and Ludwig9, Reference Yamada, Chen, Rozen and Mathews10. The functional impact of the A1298C polymorphism is less clearReference Sharp and Little5, Reference Brockton11. In vitro enzyme activity appears to be reduced in homozygous variants and compound heterozygotesReference Yamada, Chen, Rozen and Mathews10, Reference van der Put, Gabreels, Stevens, Smeitink, Trijbels, Eskes, van den Heuvel and Blom12, Reference Chango, Boisson and Barbe13, but the in vivo effects remain to be determined.
Most, but not all, previous studies have found reduced colorectal cancer risk in homozygotes for the variant C677T alleleReference Sharp and Little5, Reference Hubner and Houlston14. A1298C has been less extensively investigated, and results have been inconsistentReference Sharp and Little5, Reference Huang, Han, Li, Mao and Xie15. The inconsistencies may be due, in part, to differences in genotype distributions and folate intakes between populations.
The Scottish diet is distinguished by low vegetable intake16. This suggests folate levels may be relatively low, a view confirmed by the National Food Survey which revealed geographical variations in blood folate across Britain, with the lowest levels in ScotlandReference Bates, Mansoor, van der Pols, Prentice, Cole and Finch17. The high alcohol intakeReference Erens, Shaw, McMunn and Fields18 and smoking prevalenceReference Boreham, Shaw, McMunn and Fields19 may further compromise folate and, more generally, methyl status in the Scottish population. Furthermore, colorectal cancer incidence in Scotland is in the upper quarter of rates observed worldwideReference Parkin, Whelan, Ferlay, Teppo and Thomas20.
We undertook a population-based case–control study in the north-east of Scotland to investigate whether intakes of folate and related dietary factors, and MTHFR polymorphisms, were associated with colorectal cancer risk in a population with a relatively low folate intake.
Materials and methods
Ascertainment of cases and controls
Eligible cases were resident in the Grampian Health Board region and diagnosed with a histological confirmed first primary invasive tumour of the colon or rectum between September 1998 and February 2000. They were identified each month from the computerised records of Aberdeen Royal Infirmary pathology laboratory, which provides the centralised service for Grampian.
Controls were selected from the Grampian Community Health Index, an inventory of everyone registered with a general practitioner. The Grampian Community Health Index has a high level of completeness for the Grampian populationReference Garten, Abdalla, Reid and Russell21. Each month, a pool of potential controls was selected at random, frequency matched to cases on age and sex. Individuals who declined to take part were replaced.
The study was approved by the Joint Ethical Committee of the University of Aberdeen and Grampian Health Board (Scotland, UK). All participants gave informed consent.
Assessment of diet and other exposures
With the permission of their general practitioner, subjects were recruited by post. They completed a questionnaire comprising a 150-item semi-quantitative FFQ22 plus questions on a range of socio-demographic and lifestyle factors relevant to colorectal cancer aetiology. The FFQ had been validated in the local populationReference Masson, McNeill, Tomany, Simpson, Peace, Wei, Grubb and Bolton-Smith23 and included questions on dietary supplement use in the reference period (approximately 1 year before completion).
Genotyping
Subjects provided a mouthwash sample. DNA was extracted from exfoliated buccal cells using the Elucigene CF12 protocol (Zeneca Diagnostics, Abingdon, Oxon, UK) and amplified by PCR. The amplification products were digested with HinfI for C677TReference Frosst, Blom and Milos7 and MboII for A1298CReference van der Put and Blom24. Resultant products were separated by electrophoresis through 3 % (Metaphor®, FMC BioProducts) gels. Bands were visualised by ethidium bromide staining and UV transillumination.
Analyses were blind to case–control status. Each batch contained a negative (no template DNA) and two positive controls. Analyses were repeated if either amplification or digestion failed. Gels were double-read by two individuals and assays repeated for any samples considered ambiguous. Genotyping was also repeated, blind to the original results, for a 10 % random selection (n 67) of samples; no differences were found compared with the originally assigned genotypes.
Statistical analysis
The dietary data were converted into estimated nutrient intakes using the UK national food composition tables, taking account of cereal fortificationReference Holland, McCance and Widdowson25. For seven subjects (three cases and four controls) the FFQ was not completed or was very incomplete. A further twelve subjects (six cases and six controls) were excluded on the basis of implausible total energy intakes ( < 3347 kJ/d for men or < 2092 kJ/d for womenReference Willett26 or more than three standard deviations above mean intake in men and women separately). Protein intake was used as a surrogate for methionine. Dietary intakes were adjusted for total energy using the nutrient residual methodReference Willett26. For folate, vitamin B6, vitamin B12 and riboflavin, the primary analyses related to total intake, i.e. the sum of dietary (energy adjusted) and supplement intakes; secondary analyses related to dietary intake. Subjects were grouped into intake tertiles (for alcohol) or quartiles (other variables), based on the combined case and control distributionReference Hseih, Maisonneuve, Boyle, Macfarlane and Robertson27, with the baseline group comprising non-consumers (alcohol) or the lowest quartile. For methyl content, the ‘low’ group comprised low total folate and protein ( < median) and high alcohol (upper two tertiles) and the ‘high’ group had high total folate and protein ( ≥ median) and low alcohol (no intake or lowest tertile).
For C677T and A1298C, separate and compound genotype was assigned. Hardy–Weinberg equilibrium was assessed in controls using Pearson's χ2. Association analyses were done for individual genotypes, and combining carriers of the variant alleles, taking homozygotes for the common allele as the reference group. In addition, to explore the impact of one variant in the absence of the other, risk estimates were computed for (i) C677T genotype among 1298AA carriers and (ii) A1298C genotypes among 677CC carriers. Logistic regression was used to compute adjusted and multivariate odds ratios (OR) and 95 % CI in Stata 8 (StataCorp LP, College Station, TX, USA). Adjusted OR were adjusted for sex, age and, for the dietary variables, total energy intake. Multivariate OR were further adjusted for potential confounders; factors which made a significant contribution (likelihood ratio test P < 0·1) were retained in the model. Trend testsReference Breslow and Day28 were used to investigate dose–response across dietary quantiles or by number of variant alleles. Effect modification was explored by computing OR for combinations of (i) intake variables and (ii) intake and genotype. The test for interaction was the change in deviance ( − 2 × log likelihood) between a main-effects model and one including a cross-product term. All multivariate models were checked for adequate fitReference Hosmer and Lemeshow29.
Results
A total of 264 cases (62 % of those eligible) and 408 controls (61 %) participated. Of the 264 cases, 189 cases had colon cancer and seventy-five rectal cancer (Table 1). Cases tended to be older than controls, and a higher proportion were men.
Table 1 Characteristics of study participants by case–control status
(Numbers and percentages)
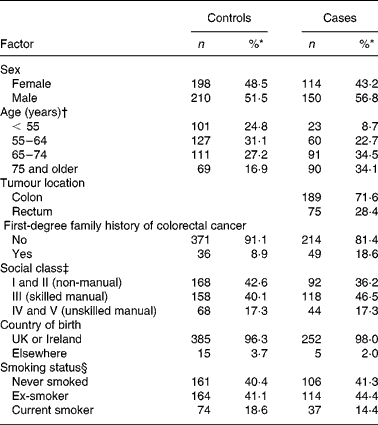
* Percentage of those subjects with complete data for a particular factor.
† Age at diagnosis for cases and at pseudo date of diagnosis for controls (middle of month in which cases diagnosed).
‡ Based on occupation at time questionnaire completed, or last occupation before retirement: for men and single women, based on own occupation; for women who were married, living as married, or widowed, based on occupation of husband or partner.
§ At approximately 1 year before the completion of the questionnaire.
Median total folate intake was 299·7 (interquartile range 263·9–348·6) μg/d (Table 2). Median dietary folate was 295 (interquartile range 261–332) μg/d. Of the subjects, 9·5 % (thirty-eight controls (9·4 %) and twenty-five cases (9·6 %)) reported taking a supplement that contained folic acid. The majority (fifty-three out of sixty-three) took supplements that included 200 μg folic acid.
Table 2 Association between intake variables and colorectal cancer
(Numbers and percentages of subjects, P values for likelihood ratio tests, odds ratios and 95 % confidence intervals)

Q, quartile; T, tertile; NSAID, non-steroidal anti-inflammatory drugs.
* Adjusted for age, sex and total energy.
† All models adjusted for sex, age, total energy, physical activity, family history of colorectal cancer, regular use of any NSAID, sex × NSAID interaction term; model for protein also adjusted for type of dietary supplement; model for alcohol also adjusted for type of dietary supplement and protein.
‡ Protein from food only.
§ High methyl content is high total folate, high protein and either no alcohol intake or intake in T1; low methyl status is low total folate, low protein and alcohol in upper two tertiles; intermediate is all other combinations of total folate, protein and alcohol. High folate or protein is intake in upper two quartiles; low folate or protein is intake in lower two quartiles.
For total folate intake, the multivariate OR exceeded unity for quartiles 2–4, but there was no clear trend (Table 2). A similar pattern was observed for total riboflavin. There was a borderline significant association between protein intake and risk (global P = 0·055; P(linear trend) = 0·066), due to a one-third reduction in risk in the highest intake group. Compared with non-drinkers, disease risk was slightly, but non-significantly, raised for all categories of alcohol intake. The association was strongest in males (multivariate OR for T1 v. none = 1·58 (95 % CI 0·60, 4·15); OR for T2 v. none = 1·57 (95 % CI 0·65, 3·80); OR for T3 v. none = 1·72 (95 % CI 0·73, 4·04)). There was a suggestion of increasing risk with decreasing dietary methyl content, although the risk estimates were not statistically significant. There were no associations between total vitamin B12 or vitamin B6 intakes and colorectal cancer.
Results of analyses restricted to dietary intake of folate, vitamin B12, vitamin B6 and riboflavin were similar to those for total intake, with slightly attenuated risk estimates (data not shown).
There was no evidence of interactions between total folate intake and total intake of vitamin B12, vitamin B6 or riboflavin, nor did particular combinations of alcohol and total folate influence risk (data not shown). Results were unchanged when analyses were repeated for dietary intake (data not shown).
The C677T genotype distribution among controls conformed with Hardy–Weinberg equilibrium (χ21 = 0·00; P = 0·999), but the A1298C distribution did not (χ21 = 6·25; P < 0·05); there was an excess of homozygous subjects (AA and CC) and a deficit of heterozygotes. For C677T, risk decreased with increasing number of variant alleles (multivariate OR for CT v. CC = 0·77 (95 % CI 0·52, 1·16); OR for TT v. CC = 0·62 (95 % CI 0·31, 1·24)) but not significantly (P(linear trend) = 0·103; Table 3). For A1298C, the risk for homozygous variant subjects was modestly, but not significantly, reduced (OR for CC v. AA = 0·81 (95 % CI 0·45, 1·49)). As regards compound genotype, no subjects were homozygous for both variants, while four cases (1·7 %) and eleven controls (2·8 %) had three variant alleles. Compared with those with no variant alleles, individuals who were double heterozygous (OR = 0·85 (95 % CI 0·41, 1·75)) or homozygous variant (OR = 0·71 (95 % CI 0·35, 1·45)) had slightly reduced risk. Among individuals who were 1298AA, multivariate OR for the C677T genotypes were: 1·00, 0·82 (95 %CI 0·40, 1·67) and 0·77 (95 % CI 0·30, 1·93). Multivariate risk estimates for the A1298C genotypes among individuals who were 677CC were: 1·00, 1·41 (95 % CI 0·67, 2·96) and 0·75 (95 % CI 0·33, 1·73).
Table 3 Association between methylenetetrahydrofolate reductase (MTHFR) genotype and colorectal cancer
(Numbers and percentages of subjects, P values for likelihood ratio tests, odds ratios and 95 % confidence intervals)

NSAID, non-steroidal anti-inflammatory drugs.
* Adjusted for age and sex.
† Adjusted for sex, age, family history of colorectal cancer, physical activity, regular use of any NSAID, sex × NSAID interaction term, total energy intake and type of dietary supplements.
‡ Three cases did not provide a mouthwash sample; genotyping failed for ten cases and fourteen controls.
§ OR for carriers of variant allele v. homozygous wild-types.
‖ Three cases did not provide a mouthwash sample; genotyping failed for sixteen cases and fourteen controls.
¶ Three cases did not provide a mouthwash sample; compound genotype could not be assigned for nineteen cases and twenty-one controls because genotyping for either C677T or A1298C had failed.
** Includes homozygous wild type for both polymorphisms or heterozygote for either polymorphism.
†† Includes heterozygotes for both polymorphisms or homozygous variant for either; OR for two or more variant alleles v. zero or one variant allele.
There was a significant interaction between C677T and total folate (Table 4; P(interaction) = 0·029). Compared with CC individuals with low ( < median) intake, risk was significantly reduced in carriers of the variant allele with low intake (OR = 0·47 (95 % CI 0·27, 0·83)), with a less pronounced risk reduction in those with high intake. A significant interaction was found between total vitamin B6 intake and C677T (P(interaction) = 0·016), which followed a similar pattern to that for total folate. Repeating the analyses for dietary folate and vitamin B6 gave less pronounced risk estimates (data not shown).
Table 4 Interactions between methylenetetrahydrofolate reductase (MTHFR) C677T genotype and dietary variables*
(Odds ratios, 95 % confidence intervals, and P values for tests for interaction)

NSAID, non-steroidal anti-inflammatory drugs.
* Folate, vitamin B12, vitamin B6, riboflavin and protein categorised at median intake; low alcohol intake is no intake or lowest tertile while high intake is intake in upper two tertiles; high methyl content is high total folate, high protein and either no alcohol intake or intake in first tertile, low methyl status is low total folate, low protein and alcohol in upper two tertiles, intermediate is all other combinations of total folate, protein and alcohol; protein based on intake from food sources.
† Adjusted for age, sex and total energy intake.
‡ All models adjusted for age, sex, total energy intake, family history of colorectal cancer, physical activity, regular use of any NSAID; model for protein also adjusted for type of dietary supplements; models for alcohol and methyl content also adjusted for type of dietary supplements and sex × NSAID interaction term.
There was a suggestion that C677T modified the associations between protein and alcohol intakes and colorectal cancer, although the tests for interaction were not statistically significant. There were no interactions between C677T and either vitamin B12 or riboflavin.
A significant interaction was observed between total folate and A1298C (Table 5; P(interaction) = 0·025). The group at lowest risk comprised homozygotes for the wild-type allele with low intake. Compared with this group, those with the AC/CC genotype and low intake or AA genotype and high intake had significantly raised risk (OR = 1·75 (95 % CI 1·00, 3·10) and OR = 1·91 (95 % CI 1·05, 3·50) respectively). Similar patterns were apparent for dietary folate (data not shown; P(interaction) = 0·060) and total (Table 5; P(interaction) = 0·033) and dietary vitamin B6 (data not shown; P(interaction) = 0·053). A1298C did not modify the relationships between colorectal cancer and vitamin B12 or riboflavin, either for total (Table 5) or dietary intake (data not shown). There were no significant interactions between A1298C and protein or alcohol.
Table 5 Interactions between methylenetetrahydrofolate reductase (MTHFR) A1298C genotype and dietary variables*
(Odds ratios, 95 % confidence intervals, and P values for tests for interaction)

NSAID, non-steroidal anti-inflammatory drugs.
* Folate, vitamin B12, vitamin B6, riboflavin and protein categorised at median intake; low alcohol intake is no intake or lowest tertile while high intake is intake in upper two tertiles; high methyl content is high total folate, high protein and either no alcohol intake or intake in first tertile, low methyl content is low total folate, low protein and alcohol in upper two tertiles, intermediate is all other combinations of total folate, protein and alcohol; protein based on intake from food sources.
† Adjusted for age, sex and total energy intake.
‡ All models (except methyl content) adjusted for sex, age, total energy, family history of colorectal cancer, physical activity, regular use of any NSAID and sex × NSAID interaction term; models for protein and alcohol also adjusted for type of dietary supplements; model for methyl content adjusted for sex, age, total energy, family history of colorectal cancer, physical activity, regular use of any NSAID and type of dietary supplements.
Discussion
Strengths and limitations
The major strength of the study was the population basis, with both cases and control recruited from population-based sampling frames. The participation rate was similar to another UK study in which contact was made by postReference Macfarlane, Gray, Kincey and Worthington30. Other than refusal, the main reasons for non-participation among cases were death (n 37; 8·7 % of those eligible) or general practitioner refusal (n 15; 3·5 %), primarily because subjects were deemed to be too ill to approach. For controls, the general practitioner refused permission to approach sixteen individuals and a further eight had died (3·6 % in total). Eligible male cases were significantly more likely to participate than females; controls resident in urban areas were significantly less likely to take partReference Sharp31. Deprivation category of residence was not significantly associated with participation among either cases or controls. Analysis of known colorectal cancer risk factors resulted in associations consistent with previous evidenceReference Little, Sharp, Masson, Brockton, Cotton, Haites and Cassidy32.
Of the controls, 12 % were 677TT homozygotes, consistent with the frequency in other white and northern European populationsReference Botto and Yang33. Of the controls, 15 % carried the 1298 CC genotype, slightly higher than the prevalence in most European studiesReference Sharp and Little5, but consistent with another study in the same area (18 % CC)Reference Sharp, Little, Schofield, Pavlidou, Cotton, Miedzybrodzka, Baird, Haites, Heys and Grubb34. The A1298C genotype frequencies were not in Hardy–Weinberg equilibrium. Since subjects were unaware of the study hypotheses or their genotype, differential participation by genotype seems unlikely. Moreover, the genotyping followed rigorous quality-control measures, which should help guard against systematic errorsReference Little, Bradley and Bray35. Other potential explanations for a departure from Hardy–Weinberg equilibrium include non-random mating, genetic drift and chance.
The FFQ was developed and extensively validated in the local area. For folate, alcohol and riboflavin, high levels of agreement (rank correlation coefficients 0·55–0·79) were found when comparing questionnaire responses with 4 d weighed recordsReference Masson, McNeill, Tomany, Simpson, Peace, Wei, Grubb and Bolton-Smith23.
The case–control design is potentially susceptible to the effects of recall bias in the assessment of lifestyle (but not genetic) risk factors. Other than this, the main limitation was limited power both for main effects and, in particular, for interactions. As regards main effects, for a genetic variant occurring in 12 % of the population (i.e. MTHFR TT) the study had 80 % power to detect an OR of 0·45 or less (α = 0·05; two-sided test); this OR is in the region of the risk estimates reported in previous studiesReference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36. It should be borne in mind that a relatively large number of statistical tests were conducted, and a proportion of positive results would be expected by chance.
Ethnic group was not assessed, but only 3 % of subjects were born outside the UK or Ireland (78 % born in Scotland; 17 % elsewhere in the UK or Ireland). In the 2001 census for Scotland, 98 % of the population described themselves as ‘white’ (Scottish, British, Irish or other) and 2 % as south-east Asian, Chinese or black37. Thus it seems unlikely that the results were adversely affected by population stratification, which needs to be quite extreme to have a major impactReference Khlat, Cazes, Genin and Guiguet38.
Folate and other intake variables
Unlike most previous studiesReference Sanjoaquin, Allen, Couto, Roddam and Key3, we did not find an inverse relationship between total folate intake and colorectal cancer. In contrast, compared with the lowest intake quartile, risk estimates for the other quartiles exceeded unity, and followed a bell-shaped pattern. A similar pattern has been reported between plasma folate and colorectal cancer in a prospective study from SwedenReference Van Guelpen, Hultdin, Johansson, Hallmans, Stenling, Riboli, Winkvist and Palmqvist39. When our analysis was restricted to subjects who did not use folic acid-containing supplements, the pattern persisted (multivariate OR: 1·0, 1·86 (95 % CI 1·07, 3·22), 1·82 (95 % CI 1·03, 3·19) and 0·96 (95 % CI 0·51, 1·79)). Although some previous studies have found the inverse relationship to be stronger for colon than rectal tumoursReference Wei, Giovannucci, Wu, Rosner, Fuchs, Willett and Colditz40, Reference Larsson, Giovannucci and Wolk41, when we stratified by site, there was no clear evidence of a trend with colon or rectal cancer. Although there may be substantial error in assessment of dietary folate intakesReference Herbert42 and supplement useReference Yang and Erickson43, the resulting misclassification is most probably random, and would be unlikely to produce the observed result.
In further analyses, in women we found a modest, albeit non-significant, reduced risk in the highest total folate group (age- and sex-adjusted OR for quartile 4 v. quartile 1 = 0·73 (95 % CI 0·35, 1·52)). Several other case–control studies found that the association between colorectal neoplasia and folate (as intake or from supplements) was only evident, or was stronger, amongst femalesReference Freudenheim, Graham, Marshall, Haughey, Cholewinski and Wilkinson44–Reference Slattery, Schaffer, Edwards, Ma and Potter48. Folic acid supplement use is reported to be higher in womenReference White, Shannon and Patterson49. In the present study, 11·6 % of females and 7·6 % of males used folic acid-containing supplements. These findings raise the possibility that folate status (or bioavailable folate, at least) may be higher for females than males for the same intake; we are not, however, aware of any evidence confirming this. There may also be a differential effect by sex on folate status of other factors influencing dietary methyl content. For example, men are more likely to drink alcohol, and to consume greater quantities, than womenReference Slattery, Schaffer, Edwards, Ma and Potter48; in the present study, 74 % of females and 86 % of males consumed alcohol. Similarly, methionine intake can be very different, possibly because men are likely to eat greater quantities of red meatReference Giovannucci, Rimm, Ascherio, Stampfer, Colditz and Willett50. Alternatively, folate intake estimates among women may be subject to less measurement error. In the validation of the FFQ used in the present study, for most nutrients the agreement between questionnaire estimates and weighed records was higher for females than malesReference Masson, McNeill, Tomany, Simpson, Peace, Wei, Grubb and Bolton-Smith23.
The median dietary folate intake among controls (295 μg/d) was close to population estimates (average intake from food and drink 274 μg/d51). In the USA, by contrast, a large proportion of the population meets the recommended intake of 400 μg/dReference Bailey52. The proportion of controls taking folic acid-containing supplements ( < 10 %) is typical of the UK populationReference Finch, Doyle, Lowe, Bates, Prentice, Smithers and Clarke53, but considerably lower than in the USAReference Slattery, Schaffer, Edwards, Ma and Potter48, Reference White, Shannon and Patterson49, Reference Keku, Millikan, Worley, Winkel, Eaton, Biscocho, Martin and Sandler54. The range of total intake in the present study was relatively narrow; cut-off points for the lowest and highest quartiles were 264 and 349 μg/d. This contrasts with several previous studies in which the cut-off point for the highest quantile was > 600 μg/dReference Giovannucci, Rimm, Ascherio, Stampfer, Colditz and Willett50, Reference Giovannucci, Stampfer, Colditz, Hunter, Fuchs, Rosner, Speizer and Willett55–Reference Harnack, Jacobs, Nicodemus, Lazovich, Anderson and Folsom57. Moreover, in most previous studies, including those where average intake was lower than in the USAReference La Vecchia, Negri, Pelucchi and Franceschi58, Reference Terry, Jain, Miller, Howe and Rohan59, there was greater variability in intake than in the present study. It is noteworthy that in the prospective study in Sweden which reported a bell-shaped relationship between plasma folate and colorectal cancerReference Van Guelpen, Hultdin, Johansson, Hallmans, Stenling, Riboli, Winkvist and Palmqvist39 – similar to our finding for folate intake – the plasma folate concentrations were considerably lower than in prospective studies from the USA that found inverse relationships with plasma or serum folateReference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36, Reference Kato, Dnistrian, Schwartz, Toniolo, Koenig, Shore, Akhmedkhanov, Zeleniuch-Jacquotte and Riboli56. Van Guelpen et al. concluded that the low folate status of their study population may have precluded detection of a protective effect of high plasma folate concentrationsReference Van Guelpen, Hultdin, Johansson, Hallmans, Stenling, Riboli, Winkvist and Palmqvist39. A similar conclusion holds for the present study (i.e. the generally low intake, and narrow range, meant that a protective effect of higher intakes could not be found). Further, in view of the weight of evidence in this area, it seems most likely that the increased risk observed for intermediate intakes is due to chance.
The lack of statistical interaction between folate and alcohol is compatible with most previous studiesReference Harnack, Jacobs, Nicodemus, Lazovich, Anderson and Folsom57–Reference Su and Arab60. Although some studies have suggested that the risk reduction associated with high folate intake was confined to those with low methionine intakeReference Giovannucci, Stampfer, Colditz, Hunter, Fuchs, Rosner, Speizer and Willett55, Reference Terry, Jain, Miller, Howe and Rohan59, we found no interaction between folate and protein (as a marker for methionine). There is tension in the previous evidence, however, in that the pattern of the folate–methionine interaction is not consistent with what would be expected on the basis of the low-methyl diet hypothesis: according to this, the combination of low intakes of folate and methionine together with high alcohol should increase disease risk. The modest raised risk we observed for intermediate and low dietary methyl content is broadly consistent with previous studiesReference Slattery, Schaffer, Edwards, Ma and Potter48, Reference Giovannucci, Rimm, Ascherio, Stampfer, Colditz and Willett50, Reference La Vecchia, Negri, Pelucchi and Franceschi58, Reference Su and Arab60. The extent to which the effect of the ‘methyl diet’ on disease risk exceeds that of folate alone remains unclear, however.
Effects of genotype
In most previous studies, carriers of three variant alleles, if they have occurred at all, have been rareReference Sharp and Little5. Our frequencies of 2·8 % among controls and 1·7 % among cases are quite high but are, of course, based on very small numbers of individuals. The high frequency, which appears to occur mainly in UK and Canadian populations, may be due to a founder effectReference Ogino and Wilson61.
The moderate risk reduction, and dose–response, associated with the 677T allele, although not statistically significant, is compatible with most previous studiesReference Sharp and Little5, Reference Hubner and Houlston14. As regards A1298C, eight of eleven studies found lower risk for the variant alleleReference Huang, Han, Li, Mao and Xie15, although not all reached significance and an effect was not seen in all subgroups. Our modest, non-significant, reduced risk in CC subjects is consistent with this.
In common with previous investigatorsReference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36, Reference Chen, Giovannucci, Kelsey, Rimm, Stampfer, Colditz, Spiegelman, Willett and Hunter62, Reference Le Marchand, Donlon, Hankin, Kolonel, Wilkens and Seifried63, we found a significant interaction between folate and C677T. However, the pattern of interaction differed from that reported previously due, in part, to differences in the folate main effect. In addition, for reasons of statistical power, we combined CT and TT genotypes in the analysis. This would have concealed the low risk in TT subjects with high folate (or dietary methyl content) that has been reported elsewhereReference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36, Reference Chen, Giovannucci, Kelsey, Rimm, Stampfer, Colditz, Spiegelman, Willett and Hunter62, Reference Slattery, Potter, Samowitz, Schaffe and Leppert64. Although we observed a significant folate–A1298C interaction, previous studies have been inconsistentReference Keku, Millikan, Worley, Winkel, Eaton, Biscocho, Martin and Sandler54, Reference Le Marchand, Donlon, Hankin, Kolonel, Wilkens and Seifried63, Reference Chen, Ma, Stampfer, Palomeque, Selhub and Hunter65. There is linkage disequilibrium between C677T and A1298C. In the present study, sixty of the sixty-eight individuals with the 677TT genotype also carried 1298AA, while seventy-nine of the eighty-seven who were 1298CC also carried 677CC. This meant that there was considerable overlap between the sub-groups at lowest risk in the two interaction analyses (C677T: low folate and CT/TT genotype; A1298C: low folate and AA genotype), which explains, in part, the different patterns of interaction observed for the two polymorphisms. More generally, differences in patterns of linkage disequilibrium between populations may contribute to heterogeneity between studiesReference Little, Sharp, Duthie and Narayanan66.
The less consistent findings for A1298C, than C677T, regarding colorectal cancer could also be because A1298C appears to have less strong functional effects, although it should be noted that the functional evidence is limited and the in vitro and in vivo impact of the variant allele is not fully resolvedReference Sharp and Little5, Reference Brockton11. In analyses of 199 controls from the present study, plasma and erythrocyte folate (measured by the microbiological assay) levels were significantly lower, and plasma homocysteine levels significantly higher, in 677TT than CC individuals, while the A1298C variant was not associated with folate or homocysteine levelsReference Narayanan, McConnell, Little, Sharp, Piyathilake, Powers, Basten and Duthie67. Neither polymorphism was related to levels of various biomarkers of DNA stability measured in lymphocytes, including strand breaks, misincorporated uracil or global methylation. The evidence from other similar studies on MTHFR and DNA stability is inconsistentReference Crott, Mashiyama, Ames and Fenech68–Reference Friso, Girelli, Trabetti, Olivieri, Guarini, Pignatti, Corrocher and Choi72. One reason for this may be that these studies, and those of intake, are not assessing folate status in the relevant tissue. What may be important is localised (rather than systemic) folate depletion acting in concert with C677TReference Brockton11. Current knowledge on folate status in colonic tissue is limited. For example, the extent to which colonic folate concentrations vary between individuals, or along the colon within a single individual, or correlate with levels in lymphocytes or dietary intakes, is not clear.
The significant interactions between vitamin B6 and MTHFR genotype followed a similar pattern and had risk estimates similar to those for folate. This may reflect the fact that dietary sources for the two nutrients overlap substantially; after adjusting for total energy intake the correlation between folate and vitamin B6 exceeded 0·70.
Some investigators have reported an alcohol–C677T interaction, such that higher alcohol intake abolished the reduced disease risk associated with the TT genotypeReference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36, Reference Keku, Millikan, Worley, Winkel, Eaton, Biscocho, Martin and Sandler54, Reference Chen, Giovannucci, Kelsey, Rimm, Stampfer, Colditz, Spiegelman, Willett and Hunter62. The present results suggested effect modification, but were not significant. The median alcohol intake among consumers was lower than the ‘high’ intake groups in previous studies (present study, 3·9 g/d (about 0·5 units/d); Chen et al. Reference Chen, Giovannucci, Kelsey, Rimm, Stampfer, Colditz, Spiegelman, Willett and Hunter62, ≥ five drinks/week; Ma et al. Reference Ma, Stampfer, Giovannucci, Artigas, Hunter, Fuchs, Willett, Selhub, Hennekens and Rozen36, ≥ one drink/d).
Most previous studies of folate and folate–MTHFR interactions have been conducted in populations where intake is relatively high, and a substantial proportion comes in the form of folic acid. The results of the present study, undertaken in a population with relatively low folate intake, suggest that the role of folate metabolism in colorectal cancer aetiology may be even more complex than previously thought. The challenge now is to unravel this complexity. 5,10-MTHFR irreversibly converts 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, the former vital for DNA synthesis and the latter key in DNA methylation. Thus, the C677T polymorphism alters the distribution of erythrocyte folatesReference Bagley and Selhub73, suggesting that particular folate species might be important in colorectal cancer. The direction of the MTHFR–colorectal cancer association – which is the opposite of that expected a priori Reference Sharp and Little5 – suggests that the availability of tetrahydrofolate and 5,10-methylenetetrahydrofolate may be particularly pertinent. This is supported by a study in colorectal tumour tissue, which found that 677TT patients had significantly lower concentrations of tetrahydrofolate and 5,10-methylenetetrahydrofolateReference Kawakami, Ruszkiewicz, Bennett, Moore, Watanabe and Iacopetta74. Expansion of the evidence on individual folate species with regard to: (1) distribution in the diet, blood and other bodily tissues in different populations; (2) factors that influence intracellular distribution and availability; and (3) disease risk, might help advance understanding of folate metabolism in colorectal neoplasia and, ultimately, shed light on carcinogenesis pathways.
Acknowledgements
The present study was funded by a grant from the National Hospitals Trust. We thank Janet Kyle, Margaret Smith, Martine Barnes and Jennifer Grzybowski for assistance with data entry and David Grubb for processing the FFQ. Graeme McHardy and Diane Thom kindly provided downloads of cases and controls. Rachel Melvin, Joanne Tomany and Mhairi Gilmour assisted with compiling data on the content of dietary supplements.





