


n-3 PUFA are essential dietary lipids receiving considerable attention as either potential nutraceuticals or protective factors for prevalent diseases. Physiologically relevant n-3 PUFA include α-linolenic acid (LNA; 18 : 3n-3 PUFA), DHA (22 : 6n-3 PUFA) and EPA (20 : 5n-3 PUFA). Unlike EPA, dietary DHA has been shown to concentrate specifically in phospholipids of brain cells, while LNA is virtually absent from nervous tissue(Reference Salem, Spiller and Scala1–Reference Umhau, Zhou and Carson3). Importantly, it is well known that high dietary consumption of DHA quickly translates into higher brain concentration in mammals(Reference Farquharson, Cockburn and Patrick4–Reference Bousquet, Saint-Pierre and Julien8). Besides their well-accepted lowering effect on plasma TAG and on the risk of CHD(Reference Harris9), preclinical, observational and intervention studies suggest that n-3 PUFA may exert beneficial effects on cognitive/memory performances(Reference Carrie, Smirnova and Clement10, Reference Fedorova, Hussein and Baumann11) and, possibly, mood disorders(Reference Kidd12), as well as Parkinson's(Reference Bousquet, Saint-Pierre and Julien8) and Alzheimer's diseases(Reference Calon and Cole13–Reference Yurko-Mauro, McCarthy and Rom16). The potential bioactivities of DHA in the brain are numerous and complex. For example, DHA has been reported to regulate membrane properties such as fluidity, elasticity and rigidity(Reference Bruno, Koeppe and Andersen17, Reference Wassall and Stillwell18) and to influence cell signalling(Reference Calon, Lim and Yang19, Reference Akbar, Calderon and Wen20), gene expression(Reference de Urquiza, Liu and Sjoberg21, Reference Ethier, Kagechika and Shudo22), neuroinflammation(Reference Calder23–Reference Lalancette-Hébert, Julien and Cordeau25) and synaptic health(Reference Calon, Lim and Morihara7, Reference Calon, Lim and Yang19). More specifically, DHA is thought to play an important role in synapse health by modulating key dendrite proteins, like drebrin and syntaxin-3. The association of drebrin with actin filaments is critical for the stability and plasticity of dendritic spines(Reference Biou, Brinkhaus and Malenka26), and DHA intake prevents drebrin loss in the Tg2576 mouse model of Alzheimer's disease(Reference Calon, Lim and Yang19). Syntaxin-3 is present in synaptic membranes(Reference Curtis, Doneske and Liu27) and in neuronal growth cones(Reference Darios and Davletov28). DHA dietary supplementation elevates synaptic levels of syntaxin-3(Reference Cansev and Wurtman29), and the action of DHA on promoting neurite outgrowth and membrane expansion has been shown to depend on syntaxin-3(Reference Darios and Davletov28). In addition, we recently reported direct evidence that chronic consumption of DHA alters the basic electrophysiological properties of entorhinal cortex (EC) neurons(Reference Arsenault, Julien and Tremblay30).
In the diet, LNA is mainly obtained from oily seeds, whereas DHA and EPA are found in fish and seafood(Reference Salem, Spiller and Scala1). In addition, LNA, EPA and DHA are among the most frequently consumed nutritional supplements. Due to its highest concentration in the brain, DHA is likely to be the most neuroactive n-3 PUFA. However, it remains unclear to what extent dietary LNA, through its hepatic conversion into EPA and DHA, can ultimately achieve the same physiological effect(Reference Brenna, Salem and Sinclair2, Reference Salem, Pawlosky and Wegher31–Reference Plourde and Cunnane33). Indeed, despite the fact that the enzymatic machinery to elongate LNA into EPA/DHA is present in the mammal liver, the capacity of this process to actually produce DHA from LNA in significant amounts is disputed, particularly in humans(Reference Brenna, Salem and Sinclair2, Reference Plourde and Cunnane33, Reference Burdge and Calder34). While it has been clearly demonstrated in animal studies that a high intake of LNA increases brain DHA concentrations(Reference Brenna, Salem and Sinclair2, Reference Fedorova, Hussein and Baumann11, Reference Plourde and Cunnane33), most reports comparing DHA v. LNA intake indicate that the maximal concentration of DHA in the brain can only be achieved by including preformed DHA in the food(Reference Brenna, Salem and Sinclair2, Reference Fedorova, Hussein and Baumann11, Reference Plourde and Cunnane33). Notwithstanding these observations, the real question whether using LNA can be as efficient as DHA to achieve central nervous system (CNS) health benefit remains open and needs to be addressed because both types of n-3 PUFA are widely available in food or supplements. In addition, although the supply of LNA through vegetables appears sustainable, consumption of DHA up to current health recommendations raises legitimate concerns, given the decline in fish stocks worldwide(Reference Jenkins, Sievenpiper and Pauly35).
To determine whether LNA is equally efficient at supplying enough DHA to the CNS for its optimal functioning, we exposed C57/BL6 mice to either an n-3 PUFA-deficient diet, an LNA diet or a DHA diet. Animals were fed with the three diets, from 4 months to 14·5 months of age, and we then recorded the intrinsic and synaptic properties of EC deep-layer neurons using whole-cell patch-clamp on brain slices. For our electrophysiological analysis, we selected EC neurons because the EC-hippocampus loop plays a key role in higher cognition/memory functions(Reference Sewards and Sewards36), which, as mentioned earlier, are thought to depend on n-3 PUFA intake. EC neurons are also known to play a determining role in temporal lobe epilepsy(Reference Kumar, Jin and Buckmaster37, Reference Bragin, Sanderson and Peterson38) and are among the first neurons affected in Alzheimer's disease(Reference Braak and Braak39–Reference Duyckaerts, Delatour and Potier42).
Experimental methods
Animals and diets
The use of animals was approved by the Laval University (Quebec, Canada) Animal Ethics Committee (approval ID = 07-113 and 07-061) and all procedures were performed according to the guidelines of the Canadian Council on Animal Care. Animals were exposed to three treatments: (1) a control safflower oil-based diet (n-3 PUFA-deficient diet); (2) a rapeseed:soyabean (4:1)-based diet containing only LNA as a source of n-3 PUFA (LNA diet); (3) a fish oil-based diet containing a high amount of DHA with a lower content in EPA (DHA diet). Ingredients and fatty acid analysis of each diet are detailed in Table 1. The LNA diet contained a higher proportion of C18 : 1n-9 MUFA than the two others due to the presence of rapeseed oil. This formula is representative of human-relevant dietary sources of LNA, which generally combine MUFA with LNA. The DHA source was a high-DHA formulation (MEG-3) from Ocean Nutrition Inc. (Halifax, Canada) in which DHA and EPA are microencapsulated into gelatin beads for protection against oxidation(Reference Hogan, O'Riordan and O'Sullivan43–Reference Whelan and Rust45). MEG-3 contains a DHA:EPA ratio of at least 3:1, both in a TAG form. In the LNA diet, the daily dose of LNA was approximately 1·5 mmol/kg per d, whereas the daily dosage of DHA and EPA was 1·14 and 0·36 mmol/kg per d, respectively, in the DHA diet (assuming that a 25 g mouse eats 3 g of food/d). Only purified diets with precisely determined formulae were used in the present study to avoid any batch-to-batch variations. The three diets were isoenergetic and were exactly the same in terms of total fat (5 % w/w), total carbohydrates (66 % w/w), total protein (20 % w/w), total energy (4 kcal/g or 16·3 kJ/g), fibres, vitamins, minerals and antioxidants, and did not contain phyto-oestrogens unlike most laboratory chows (Table 1). A small difference in the total fat content (4·8 % in the DHA diet instead of 5 %) was probably due to small variation in the estimated value v. the actual measured values after incorporation of the MEG-3 formulation. The dietary treatment lasted 10 months, starting at 4 months of age and ending at killing of the animal between 14 and 15 months of age (14·5 (sem 0·5) months). This treatment period was selected to study the effects of diet in adult animals and to avoid interfering in the developmental or maturation process.
Table 1 Description of n-3 PUFA-deficient, α-linolenic acid (LNA) and DHA diets

LA, linoleic acid; AA, arachidonic acid; DTA, docosatetranoic acid; DPA, docosapentaenoic acid.
* Ocean Nutrition Inc., Halifax, Canada.
† Measured by GC.
Preparation of tissue samples
All the experiments were performed in the same animals. The right hemisphere was devoted for electrophysiology studies. The left hemisphere was quickly dissected and the frontal cortex was used for lipid measurements and the parieto-temporal cortex was assigned for Western immunoblots. It was not possible to use the same tissue for both biochemical procedures, as fatty acids (FA) are very sensitive to oxidation. However, previous analyses in rodents and non-human primates have shown that the fatty acid profile is similar between the cortical areas and are affected evenly by DHA supplementation(Reference Diau, Hsieh and Sarkadi-Nagy46–Reference Hsieh and Brenna49).
Protein extraction
After adding eight volumes of Tris-buffered saline (TBS) containing Complete™ protease inhibitors cocktail (Roche, Indianapolis, IN, USA), 10 μg/ml of pepstatin A, 0·1 mm-EDTA and phosphatase inhibitors (1 mm each of sodium vanadate and sodium pyrophosphate, 50 mm sodium fluoride), the frozen samples were sonicated briefly (3 × 10 s) and centrifuged at 100 000 g for 20 min at 4°C to generate a TBS-soluble intracellular and extracellular fraction (soluble fraction). The TBS-insoluble pellet was sonicated in five volumes of lysis buffer (150 mm-NaCl, 10 mm-NaH2PO4, 1 % Triton X-100, 0·5 % SDS and 0·5 % deoxycholate) containing the same protease and phosphatase inhibitor cocktail. The resulting homogenate was centrifuged at 100 000 g for 20 min at 4°C to produce a lysis/detergent-soluble (DS) fraction (membrane fraction).
Western immunoblotting
For Western immunoblotting, protein concentration was determined using bicinchoninic acid assays (Pierce, Rockford, IL, USA). Equal amounts of protein per sample (15 μg of total protein per lane) were added to Laemmli's loading buffer, heated to 95°C for 5 min before loading and subjected to SDS-PAGE. Proteins were electroblotted onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) before blocking in 5 % non-fat dry milk and 1 % bovine serum albumin in PBS-Tween 20 for 1 h. Membranes were immunoblotted with appropriate primary and secondary antibodies followed by chemiluminescence reagents (Lumiglo Reserve; KPL, Gaithersburg, MD, USA). Band intensities were quantified using a KODAK Image Station 4000 MM Digital Imaging System (Molecular Imaging Software version 4.0.5f7; Carestream Health, Rochester, NY, USA). The following antibodies were used in this study: anti-actin (ABM, Richmond, Canada), anti-drebrin (MBL International, Woburn, MA, USA), anti-postsynaptic density protein 95 (Millipore), anti-synaptophysin (Millipore), anti-syntaxin 3 (Sigma, St Louis, MO, USA).
Lipid extraction and GC
Lipid profiles were determined in the frontal cortex, as the fatty acid profile is reported to be similar throughout the brain cortex(Reference Diau, Hsieh and Sarkadi-Nagy46, Reference Brenna and Diau47). GC experiments were performed as previously described(Reference Bousquet, Saint-Pierre and Julien8, Reference Arsenault, Julien and Tremblay30, Reference Julien, Berthiaume and Hadj-Tahar50). Approximately 20 mg of frozen frontal cortex from each mouse were used for lipid extraction. Weighed brain tissues were homogenised with butylated hydroxytoluene-methanol (Sigma) and two volumes of CHCl3 (J.T. Baker, Phillipsburg, NJ, USA) and 0·5 volumes of NaH2PO4 (0·2 m)-buffer solution were added to the resulting homogenate. After centrifugation at 2500 g, the lower layer (CHCl3 fraction) was collected(Reference Folch, Lees and Sloane Stanley51). Lipid extracts were transmethylated with BF3–methanol (Alltech, State College, PA, USA) at 100°C for 60 min. After cooling down, 2 ml of water and 2 ml of hexane (J.T. Baker) were added. After homogenisation and centrifugation at 2500 g, the upper layer (hexane fraction) was collected, dried down to about 100 μl and transferred to a GC autosampler vial and capped under N2. Fatty acid methyl ester profiles in brain tissue were obtained by capillary GC using a temperature gradient on an HP5890 gas chromatograph (Hewlett Packard, Toronto, Canada) equipped with an HP-88 capillary column (100 m × 0·25 mm inner diameter × 0·20 μm film thickness; Agilent Technologies, Mississauga, ON, Canada) coupled with a flame ionisation detector. Helium was used as a carrier gas (split ratio 1:80). FA were identified according to their retention time, using the following standard mixtures as a basis for comparison: the FAME 37 mix (Supelco, Inc., Bellefonte, PA, USA) and the GLC-411 fatty acid mix (NuChek Prep, Inc., Elysian, MN, USA), as well as the following methylated FA C22 : 5n-6 (Larodan AB, Malmö, Sweden) and C22 : 5n-3 (Supelco, Inc.). Finally, a mixture of trans-FA containing: C18 : 2n-6 cis/trans (Supelco, Inc.), a mixture of cis/trans C18 : 3n-3 (Supelco, Inc.), the following Nucheck FA: C14 : 1 trans-9, C16 : 1 trans-9, and finally, the isoforms of C18 : 1 (cis-6, trans-6, cis-11, trans-11: Nucheck, and cis-12, cis-13; Supelco, Inc.) were also used as a standard mixture.
Slice preparation for electrophysiology recordings
Brain slices were prepared as previously described(Reference Zhang and Arsenault52, Reference Arsenault and Zhang53). Briefly, mice were deeply anaesthetised with ketamine (100 mg/kg, intraperitoneally) and xylazine (10 mg/kg, intraperitoneally), and decapitated. The brain was removed quickly (<60 s) and placed in an ice-cold solution containing (mm): 210 sucrose, 3·0 KCl, 1·0 CaCl2, 3·0 MgSO4, 1·0 NaH2PO4, 26 NaHCO3 and 10 glucose, and saturated with 95 % O2:5 % CO2. Horizontal slices of 300 μm were cut from inferior to superior brain with a vibrating tissue slicer (VT 1000 s; Leica, Wetzlar, Germany) and kept in artificial cerebral spinal fluid containing (mm): 124 NaCl, 3·0 KCl, 1·5 CaCl2, 1·3 MgCl2, 1·0 NaH2PO4, 26 NaHCO3 and 20 glucose, and saturated with 95 % O2:5 % CO2 at room temperature (21–23°C). Slices were allowed to recover for at least 1 h before recording.
Patch-clamp recording
For recording, a slice was transferred to a submerge-type chamber and continuously exposed to artificial cerebral spinal fluid heated to 30–32°C, saturated with 95 % O2:5 % CO2 and flowing at a rate of 2·0 (sem 0·2) ml/min. The slices were viewed first with a 4 × objective and a deep layer of EC was located beside the hippocampus (Fig. 1). In general, two slices can be recorded per hemisphere. Large deep-layer neurons in the EC were then viewed under near-IR illumination with a 40 × water-immersion objective (Fluor, 40 × , 0·80W; Nikon, Mississauga, Canada) and a charge-coupled device camera (IR-1000; Dage MTI, Michigan City, IN, USA).

Fig. 1 Electrophysiology methodology. (a) Preparation of slices by horizontal cutting. The black line illustrates the cutting axis. (b) Brain slice stained with haematoxylin. The recorded neurons are those of deep layers of lateral entorhinal cortex. CPu, Caudate putamen (striatum); Hipp, hippocampus; REC, recording electrode. (c) Illustration of the patch-clamp technique. The neuron is labelled with neurobiotin and revealed by immunohistology (nickel-3,3′diaminobenzidine tetrahydrochloride (DAB) revelation).
Experiments were conducted at 30–32°C. Patch pipettes were pulled from thick wall borosilicate glass (1·5/0·84 mm; WPI, Sarasota, FL, USA) on a horizontal puller (P-97; Sutter Instruments, Novato, CA, USA). The pipette solution contained (mm): 100 KMeSO4, 15 KCl, 4 ATP-Mg, 10 creatine phosphate, 10 HEPES, 0·5 ethylene glycol tetraacetic acid (pH 7·2, adjusted with KOH). Electrodes had resistances between 2 and 5 MΩ. Liquid junction potential, estimated to be 5 mV, was not corrected. The seal resistance was greater than 2 GΩ. Whole-cell recordings were made at the soma with a Multiclamp 700A amplifier (Molecular Devices, Sunnyvale, CA, USA). The serial resistance, usually between 7 and 20 MΩ, was not compensated. Experiments were conducted using the Axograph 4.9 program (Molecular Devices). Data were digitised at 8 or 16 kHz and were either filtered at 1 kHz, depending on the recording protocol.
Data analysis for electrophysiology experiments
AxoGraph 4.9 (Molecular Devices Corporation, Downingtown, PA, USA) and Origin 7 (OriginLab, Northampton, MA, USA) software programs were used for analysis. Passive properties were studied in current clamp mode, whereas spontaneous excitatory postsynaptic currents (sEPSC) were quantified in voltage-clamp mode with a voltage clamp to − 60 mV. The cell conductance (represented by G c in formula) was estimated from the slope of the graph of hyperpolarised current injection (represented by I in formula) v. voltage variation (represented by V in formula). The calculation was derived from the equation I = G c × V. The injected current duration was 400 ms and current intensities were 50, 100, 150 and 200 pA. Cell capacitance (represented by CC in formula) (i.e. for a first-order resistance-capacitance circuit) was estimated from the linear slope of the plot of I × T = CC × V, where T is the time constant of voltage variation, measured by fitting a single exponential function for a voltage decay over time, V = V ∞(1 − e − T/(R × CC)), where R is the input resistance (i.e. ![]() ) and V ∞ is the asymptote, so that T = RC (i.e. V = 0·632 V ∞), using a graphical method. CC corresponds to the linear slope of the graph displaying the relationship between I × T v. V. Passive properties were studied in current-clamp mode because this recording configuration is more accurate than voltage-clamp mode(Reference Golowasch, Thomas and Taylor54). The membrane conductance (represented by G m in formula; conductance for 1 F) was calculated from the resistance and capacitance of the neuron using the formula G m = G c/CC. The sEPSC were automatically detected using the event detection package of AxoGraph. This package uses a pre-established template (amplitude: 5 pA, rise-time: 0·6 ms, decay time: 3·5 ms, baseline: 1·5 ms, length: 4 ms) to detect synaptic events. Events with amplitude < 4 pA were rejected.
) and V ∞ is the asymptote, so that T = RC (i.e. V = 0·632 V ∞), using a graphical method. CC corresponds to the linear slope of the graph displaying the relationship between I × T v. V. Passive properties were studied in current-clamp mode because this recording configuration is more accurate than voltage-clamp mode(Reference Golowasch, Thomas and Taylor54). The membrane conductance (represented by G m in formula; conductance for 1 F) was calculated from the resistance and capacitance of the neuron using the formula G m = G c/CC. The sEPSC were automatically detected using the event detection package of AxoGraph. This package uses a pre-established template (amplitude: 5 pA, rise-time: 0·6 ms, decay time: 3·5 ms, baseline: 1·5 ms, length: 4 ms) to detect synaptic events. Events with amplitude < 4 pA were rejected.
Statistical analysis
Values were expressed as means with their standard errors. Statistical comparisons were performed using a one-way ANOVA followed by the Tukey–Kramer post hoc test. Correlation analyses were performed using linear regressions. Percentage of spontaneously active neurons was analysed by contingency tables using Pearson's χ2 test.
Results
Effects of PUFA treatment on brain fatty acid profiles
Several studies have emphasised the critical influence of dietary fat composition on the accumulation of specific FA in the brain(Reference Bousquet, Saint-Pierre and Julien8, Reference Bourre, Francois and Youyou55–Reference Green, Martinez-Coria and Khashwji57, Reference Julien, Tremblay and Phivilay58). To confirm the efficacy of our dietary treatment, we determined the fatty acid profile in the frontal cortex of each animal. The most relevant results are shown in Fig. 2, whereas the detailed data are given in Table S1 of the supplementary material (available online http://www.journals.cambridge.org/bjn). The dietary treatments did not significantly modulate total SFA (approximately 44 %) and total MUFA (approximately 21 %), but total PUFA were slightly lower following LNA intake ( − 4 to − 6 %; P < 0·05). DHA accounted for 85–88 % of cerebral n-3 PUFA, while levels of EPA remained very low and LNA was undetectable (Fig. 2 and Table S1, supplementary material for this article can be found at http://www.journals.cambridge.org/bjn). As expected, DHA consumption increased the concentrations of DHA and total n-3 PUFA in the brain compared to the other two diets (32–34 % compared to the n-3 PUFA-deficient diet and 9–11 % compared to the LNA diet; P < 0·001). Conversely, we observed a decrease of arachidonic acid (AA; C20 : 4n-6 PUFA; − 23 % compared to the n-3 PUFA-deficient diet and − 11 % compared to the LNA diet; P < 0·001) and total n-6 PUFA ( − 33 % compared to the n-3 PUFA-deficient diet and − 10 % compared to the LNA diet; P < 0·001) following consumption of DHA. LNA intake also increased DHA and n-3 PUFA (P < 0·001, 23 % compared to the n-3 PUFA-deficient diet) and decreased AA (P < 0·001, − 13 % compared to the n-3 PUFA-deficient diet) and n-6 PUFA (P < 0·001, − 25 % compared to the n-3 PUFA-deficient diet) in the cortex of mice, but to a lesser degree compared to the diet containing preformed DHA from fish oil (P < 0·001; Fig. 2). The DHA:AA ratio was thus greatly increased following DHA intake (74 %, P < 0·001), whereas the LNA diet had an intermediate effect (41 % compared to the n-3 PUFA-deficient diet, P < 0·001 v. the other two groups, Table S1, supplementary material for this article can be found at http://www.journals.cambridge.org/bjn). In addition, LNA intake increased the concentrations of C18 : 1n-9 MUFA (12 %, P < 0·05) and induced a switch from C18 : 0 to C16 : 0 (P < 0·05). Levels of docosapentaenoic acid (C22 : 5n-6 PUFA) remained low in DHA or LNA-fed animals, compared to control animals (P < 0·001). In summary, high chronic intake of DHA massively increased brain concentrations of DHA, whereas the DHA-increasing effect of LNA treatment was highly significant, but less striking.
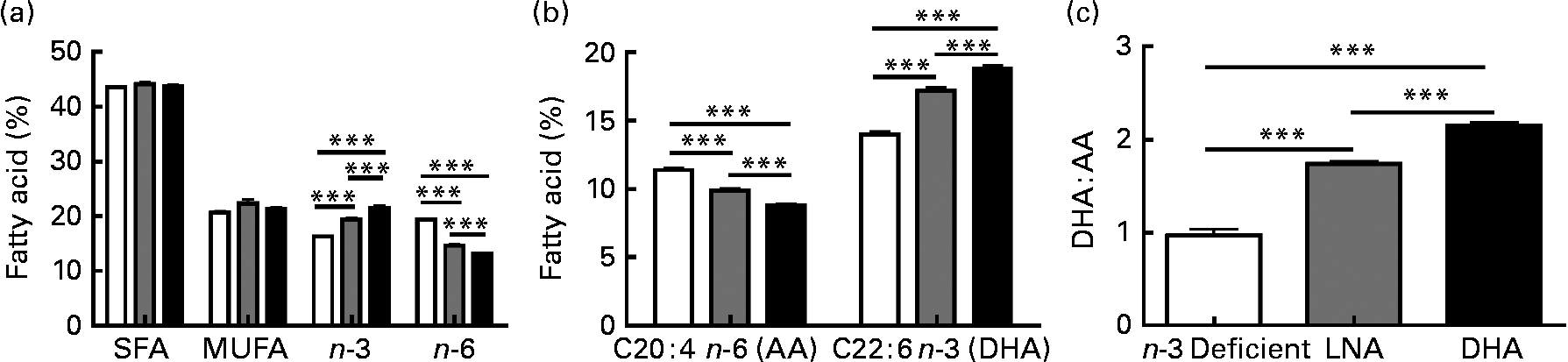
Fig. 2 Effects of α-linolenic acid (LNA, ![]() ) and DHA (■) consumption on the relative percentage of fatty acid in frontal cortex. (a) Dietary DHA increased the amount of total n-3 PUFA and decreased total n-6 PUFA compared to n-3 PUFA-deficient diet (□). The LNA-enriched diet produced an intermediate effect on n-3 PUFA and n-6 PUFA levels. SFA and MUFA levels were not modified by diets. (b) Chronic DHA intake increased the proportion of DHA and reduced the proportion of arachidonic acid (AA) in the frontal cortex, compared to n-3 PUFA-deficient diet. Again, the LNA diet produced an intermediate effect. (c) Consumption of both LNA or DHA increased the DHA:AA ratio, compared to n-3 PUFA-deficient diet, but the effect of DHA was significantly more pronounced. Values are means, with their standard errors represented by vertical bars, n 9 for the n-3 PUFA-deficient group and n 13 for LNA and DHA groups. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer post hoc test. *** Mean values were significantly different (P < 0·001).
) and DHA (■) consumption on the relative percentage of fatty acid in frontal cortex. (a) Dietary DHA increased the amount of total n-3 PUFA and decreased total n-6 PUFA compared to n-3 PUFA-deficient diet (□). The LNA-enriched diet produced an intermediate effect on n-3 PUFA and n-6 PUFA levels. SFA and MUFA levels were not modified by diets. (b) Chronic DHA intake increased the proportion of DHA and reduced the proportion of arachidonic acid (AA) in the frontal cortex, compared to n-3 PUFA-deficient diet. Again, the LNA diet produced an intermediate effect. (c) Consumption of both LNA or DHA increased the DHA:AA ratio, compared to n-3 PUFA-deficient diet, but the effect of DHA was significantly more pronounced. Values are means, with their standard errors represented by vertical bars, n 9 for the n-3 PUFA-deficient group and n 13 for LNA and DHA groups. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer post hoc test. *** Mean values were significantly different (P < 0·001).
α-Linolenic acid did not modulate passive properties of neurons: comparison with DHA
We next sought to determine how LNA treatment influenced the basic properties of neurons within the mouse EC-hippocampal circuitry, compared to DHA (Fig. 3(a) and (b)). A summary of results is presented in Fig. 3, and given in more detail in Table S2 of the supplementary material (available online at http://www.journals.cambridge.org/bjn). First, we observed that cell conductance, an indicator of the flux of ions crossing the membrane via ion channels, was higher in DHA-enriched EC neurons (P < 0·05 compared to the n-3 PUFA-deficient diet; Fig. 3(c) and (d)). Second, increased CC, an indicator of total membrane area(Reference Golowasch, Thomas and Taylor54, Reference Gentet, Stuart and Clements59, Reference Faumont, Boulin and Hobert60), was recorded in neurons from animals fed DHA compared to the n-3 PUFA-deficient diet (P < 0·05, Fig. 3(e) and (f)). A strong correlation was found between CC and cell conductance (P < 0·01, r 2 0·258, n 37). Third, cellular time constant and membrane conductance (conductance per capacitance unit) were not altered by dietary PUFA. Finally, DHA induced a hyperpolarisation of EC neurons compared to the n-3 PUFA-deficient diet (P < 0·05; Fig. 3(g)), whereas animals fed LNA had an intermediate resting potential not significantly different from the n-3 PUFA-deficient diet or DHA groups. Consequently, we found an increase of spontaneously active neurons after the n-3 PUFA-deficient diet (45 %), compared to LNA- (18 %) and DHA-rich (9 %) diets (Fig. 3(h)). Overall, LNA treatment had little effect on passive properties of EC neurons, suggesting that changes in capacitance, conductance and resting potential occurred only after the brain concentrations of DHA reached a threshold. LNA consumption was nonetheless sufficient to diminish the spontaneous activity of EC neurons.

Fig. 3 Chronic DHA intake modulated the passive properties of entorhinal cortex (EC) deep layer neurons. (a) Electrical representation of a cell membrane. (b) To quantify the passive properties, different intensities of hyperpolarising current were injected into the neuron in current clamp and the voltage variation (V) and time constant (T) were measured after each injection. (c) Relationship between the hyperpolarising current injections and resulting Vs of EC neurons of mice fed with n-3 PUFA-deficient (![]() ), α-linolenic acid (LNA, ▲) or DHA (●) diet. The slope corresponds to the cell conductance of the neuron. (d) In contrast to DHA, LNA intake did not increase the cell conductance of EC neurons, compared to n-3 PUFA-deficient group. (e) Relationship between the injected current × T and the V recorded in EC neurons of mice fed with the three different diets. The slope of each linear curve corresponds to cell capacitance. (f) In contrast to DHA, LNA intake did not increase the cell capacitance of EC neurons. (g) The membrane potential of neurons was hyperpolarised in mice fed the DHA diet, compared to n-3 PUFA-deficient diet. (h) Dietary intake of LNA or DHA decreased the number of spontaneously active EC neurons. Values are means, with their standard errors represented by vertical bars. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer post hoc test and a Pearson's χ2 test (% of neurons spontaneously active). The numbers of recorded neurons were 12, 13 and 12 for n-3 PUFA-deficient, LNA and DHA groups, respectively. * Mean values were significantly different from those of LNA diet group (P < 0·05). † Mean values were significantly different from those of n-3 PUFA-deficient group (P < 0·05).
), α-linolenic acid (LNA, ▲) or DHA (●) diet. The slope corresponds to the cell conductance of the neuron. (d) In contrast to DHA, LNA intake did not increase the cell conductance of EC neurons, compared to n-3 PUFA-deficient group. (e) Relationship between the injected current × T and the V recorded in EC neurons of mice fed with the three different diets. The slope of each linear curve corresponds to cell capacitance. (f) In contrast to DHA, LNA intake did not increase the cell capacitance of EC neurons. (g) The membrane potential of neurons was hyperpolarised in mice fed the DHA diet, compared to n-3 PUFA-deficient diet. (h) Dietary intake of LNA or DHA decreased the number of spontaneously active EC neurons. Values are means, with their standard errors represented by vertical bars. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer post hoc test and a Pearson's χ2 test (% of neurons spontaneously active). The numbers of recorded neurons were 12, 13 and 12 for n-3 PUFA-deficient, LNA and DHA groups, respectively. * Mean values were significantly different from those of LNA diet group (P < 0·05). † Mean values were significantly different from those of n-3 PUFA-deficient group (P < 0·05).
DHA, but not α-linolenic acid, increased spontaneous excitatory post-synaptic currents frequency
To probe whether LNA consumption changes the network integration of EC neurons, we recorded the sEPSC, an indicator of excitatory synaptic connectivity. We found that neurons from DHA-fed animals detected more sEPSC than those from LNA or n-3 PUFA-deficient-fed animals (P < 0·05, Fig. 4(a) and (b) and Table S2, supplementary material for this article can be found at http://www.journals.cambridge.org/bjn). However, the increase in sEPSC became undetectable when the frequency of sEPSC was normalised to CC (Fig. 4(c)), strongly suggesting that the rise in sEPSC was a consequence of increased membrane surface area of the neurons. The positive correlation between sEPSC and CC supports this hypothesis (P < 0·05, r 2 0·2031, n 26). The amplitudes of sEPSC remained unchanged between the groups (Fig. 4(d)), suggesting that a similar intensity of electrical or ion transfer was channelled through each synapse.

Fig. 4 α-Linolenic acid (LNA) consumption did not reproduce the increase in frequency of spontaneous excitatory postsynaptic current (sEPSC) observed after a DHA-enriched diet. (a) Examples of intracellular recordings of sEPSC (voltage clamped at − 60 mV). (b) Treatment with DHA increased the frequency of sEPSC. (c) No difference was found for sEPSC frequency normalised to cell capacitance, an indicator of total membrane surface. (d) sEPSC amplitude was not affected by dietary treatments. Values are means, with their standard errors represented by vertical bars. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer post hoc test. The numbers of recorded neurons were 11, 7 and 8 for n-3 PUFA deficient, LNA and DHA groups, respectively. Mean values were significantly different: * P < 0·05, ** P < 0·01.
Implication of syntaxin-3 and drebrin in the regulation of cell capacitance by DHA diet: threshold effect
The membrane expansion induced by DHA, as indicated by the increase in CC, is consistent with morphological assessments of cultured neurons exposed to DHA(Reference Robson, Dyall and Sidloff61–Reference He, Qu and Cui63). Two major molecular mechanisms particularly suspected to mediate this effect of DHA involve the synaptic proteins drebrin and syntaxin-3. The role of drebrin in the regulation of postsynaptic cytoarchitecture has been compellingly documented(Reference Shim and Lubec64, Reference Sekino, Kojima and Shirao65) and DHA has been shown to prevent the translocation of drebrin from the membrane to the cytosol in Tg2576 mice(Reference Calon, Lim and Yang19), a model of Alzheimer's disease with neuronal atrophy(Reference Moolman, Vitolo and Vonsattel66). On the other hand, the binding to syntaxin-3 of long-chain PUFA such as DHA has been identified as a critical step in neurite outgrowth(Reference Darios and Davletov28). To verify whether drebrin and syntaxin-3 were implicated here, their concentrations were quantified in cytosolic (TBS/soluble fraction) and membrane homogenate preparations (DS fractions) from the parieto-temporal cortex of mice. First, we observed that CC correlated positively with syntaxin-3 levels (Fig. 5(a)) and with the translocation ratio (membrane/cytosol: DS/TBS fractions) of drebrin (Fig. 5(b)). In addition, we noted a positive relationship between syntaxin-3 concentrations and the drebrin translocation ratio (Fig. 5(c)), suggesting a link between these two molecular pathways. Second, we found that chronic intake of preformed DHA increased syntaxin-3 levels (P < 0·05; Fig. 5(d)) and reduced the DS:TBS drebrin ratio (P < 0·05; Fig. 5(e)), compared to both diets without DHA (LNA and n-3 PUFA-deficient diets). Values of syntaxin-3 were 0·81 (sem 0·06; n 7) for no DHA group and 1·19 (sem 0·14; n 5) for DHA group, whereas they were 0·86 (sem 0·07; n 6) and 1·20 (sem 0·12; n 6) for DS:TBS ratio of drebrin, respectively (Fig. 5(d) and (e)). Thirdly, we found that changes in CC, syntaxin-3 and DS:TBS drebrin ratio were observed only when the DHA:AA ratio reached a certain level (Fig. 5(f)–(h)). Indeed, we noted that a DHA:AA ratio threshold value of ≥ 2:1 was necessary to induce an increase in CC (Fig. 5(g)), in syntaxin-3 (Fig. 5(h)) and the translocation ratio of drebrin from the membrane to the cytosol (Fig. 5(i)), as obtained only after high DHA consumption. Altogether, these corroborating observations suggest a strong association between syntaxin-3 concentrations, drebrin translocation and the DHA-induced membrane expansion, and are in accordance with a threshold effect of DHA. Furthermore, the intermediate effect of LNA diet (between n-3 PUFA-deficient and DHA diets) was not sufficient to alter any basic parameters of EC cells, except the number of spontaneously active neurons.

Fig. 5 Association between drebrin and syntaxin-3 and cell capacitance. Cell capacitance positively correlated with (a) syntaxin-3 levels (r 2 0·63, P < 0·01) and (b) detergent-soluble (DS):Tris-buffered saline (TBS) drebrin translocation ratio (r 2 0·62, P < 0·01), whereas a positive relationship was observed between (c) syntaxin-3 and the drebrin translocation ratio (r 2 0·57, P < 0·01). Dietary intake of DHA led to (d) an increase in syntaxin-3 and (e) a decrease in the drebrin translocation ratio, compared to n-3 PUFA-deprived control animals (d and e). While the DHA:arachidonic acid (AA) ratio was linearly associated with (f) resting potential (r 2 0·41, P < 0·05), (g) cell capacitance, (h) DS syntaxin-3 and (i) the DS:TBS drebrin translocation ratio were altered after the DHA:AA ratio reached approximately 2:1 in the brain. The dotted line on each graph represents the average DHA:AA ratio following each diet, whereas the horizontal bar at the end represents the standard error mean. Statistical comparisons were performed using unpaired t test (d and e) or linear regression (a, b, c, f). * Mean values were significantly different from those of animal receiving no DHA (P < 0·05). n-3 Def, n-3 PUFA deficient.
Discussion
The data generated in the present study are consistent with the following conclusions. First, chronic LNA consumption increased DHA and reduced AA concentrations in the brain cortex, but to a lesser extent than treatment with an equimolar daily dose of preformed DHA. Second, LNA treatment did not replicate the DHA-induced increases in CC and sEPSC frequencies. Third, the effect of DHA on CC was strongly associated with increased concentrations of syntaxin-3 and drebrin translocation towards the membrane fractions. The effect of brain DHA on CC, syntaxin-3 and drebrin appears to occur above a DHA:AA ratio threshold, reachable only with dietary intake of preformed DHA diet and not with intake of LNA. Finally, treatment with DHA hyperpolarised the resting potential of EC neurons and decreased the number of spontaneously active neurons, whereas LNA consumption led to an intermediate effect on these electrophysiological parameters.
DHA readily accumulates in the brain: partial conversion of α-linolenic acid into DHA
As expected, dietary consumption of preformed DHA was readily translated in the brain fatty acid profiles. Indeed, the intake of DHA during 10 months led to increased DHA concentrations in the frontal cortex, compared to animals fed the control, n-3 PUFA-deficient diet. Such observation is consistent with numerous previous reports(Reference Salem, Spiller and Scala1, Reference Calon, Lim and Morihara7, Reference Bousquet, Saint-Pierre and Julien8, Reference Arsenault, Julien and Tremblay30, Reference Brenna and Diau47) and it may be explained in part by the fact that DHA readily crosses the blood–brain barrier in vivo (Reference Ouellet, Emond and Chen67), where it stably incorporates into cerebral phospholipids(Reference Umhau, Zhou and Carson3, Reference Rapoport, Rao and Igarashi68).
The effect of LNA on brain n-3 PUFA concentration is less straightforward than preformed DHA. LNA is a metabolic precursor of DHA and mammals have the enzymatic machinery to transform LNA into DHA(Reference Brenna, Salem and Sinclair2, Reference Plourde and Cunnane33). The important relationship between dietary LNA and brain DHA has been known for decades(Reference Mohrhauer and Holman69, Reference Galli, White and Paoletti70), and has been confirmed in many animal studies(Reference Salem, Spiller and Scala1, Reference Moriguchi, Greiner and Salem6–Reference Bousquet, Saint-Pierre and Julien8, Reference Bourre71, Reference Brenna72). However, studies have also shown that this process may be limited in mammals, and almost negligible in humans exposed to usual diets(Reference Brenna, Salem and Sinclair2, Reference Plourde and Cunnane33). Here, an equimolar intake of LNA led to a significant increase in brain DHA, which did not match the effect of the high-DHA diet, in agreement with previous studies in rodents(Reference Brenna, Salem and Sinclair2, Reference Abedin, Lien and Vingrys73). Thus, our results are consistent with the contention that supplementation in preformed DHA is manifold more efficient than an equivalent daily dose of LNA to increase the concentration of DHA in the brain.
Nevertheless, we observed that both LNA and DHA reduced n-6 docosapentaenoic acid concentrations, a result consistent with previous analyses in animals(Reference Fedorova, Hussein and Baumann11, Reference Abedin, Lien and Vingrys73). Indeed, a brain n-6 docosapentaenoic acid elevation indicates a state of DHA deficiency in rodents, probably reflecting desperate attempts by the brain to generate twenty-two carbon PUFA in place of the missing DHA(Reference Salem, Moriguchi and Greiner56, Reference Youyou, Durand and Pascal74). This observation is important because it shows that LNA intake alone was efficient to fully reverse the state of DHA deprivation found in animals fed a n-3-PUFA-depleted diet. In other words, based on such data, one might expect the LNA-enriched diet to be sufficient to avoid the disruption of PUFA-related brain activities.
α-Linolenic acid diet does not achieve the effect of DHA on cell capacitance and the frequency of spontaneous excitatory postsynaptic currents
Despite the fact that the LNA-enriched diet increased DHA concentration and decreased the level of AA compared to the n-3 PUFA-deficient diet, it was apparently not sufficient to induce changes in CC and synaptic activity similar to dietary supplementation of preformed DHA. Thus, the present data suggest that LNA, the essential precursor of n-3 PUFA, can be partly converted to DHA, but in insufficient quantity to modulate the CC of EC neurons. An apparent threshold relationship between CC and DHA:AA ratio, reached exclusively with DHA intake, strongly supports this view. In addition, DHA intake modulated the brain concentrations of other bioactive PUFA, such as 22 : 5n-3 PUFA, compared to the LNA-based diet (see Table S2 of the supplementary material available online at http://www.journals.cambridge.org/bjn), which could also underlie the present electrophysiological observations. For example, 22 : 5n-3 (n-3 docosapentaenoic acid) is a known precursor of cyclo-oxygenase 2-dependent electrophilic oxo-derivatives molecules, which are potent anti-inflammatory mediators(Reference Groeger, Cipollina and Cole75).
Since the capacitance of a given membrane surface area is a constant value for all cells(Reference Gentet, Stuart and Clements59), the estimation of decay time constant of voltage variation to calculate CC directly reflects the total membrane surface(Reference Golowasch, Thomas and Taylor54, Reference Gentet, Stuart and Clements59). Accordingly, the electrophysiological quantification of CC was previously used to measure membrane atrophy during the neurodegenerative process in an Alzheimer mouse model(Reference Arsenault, Julien and Tremblay30) and to measure the membrane expansion of interneurons during development of Caenorhabditis elegans (Reference Faumont, Boulin and Hobert60). Such an enlarging effect of DHA on EC cells thus suggests that DHA intake promoted the network integration of neurons in vivo (Reference Cao, Kevala and Kim62). Supporting this idea, we found consequently a higher synaptic detection by DHA-enriched EC neurons. In accordance with our study, DHA promotes neurite outgrowth and synaptic formation in vitro and in vivo (Reference Robson, Dyall and Sidloff61, Reference Cao, Kevala and Kim62, Reference Wurtman, Cansev and Ulus76), whereas a higher dentritic spine density was reported in hippocampal neurons of gerbil following DHA and uridine-5-monophosphate supplementation(Reference Sakamoto, Cansev and Wurtman77). The role of DHA in promoting/stabilising excitatory synapses might involve inhibition of glutamate transporters located on astrocytes(Reference Susarla, Seal and Zelenaia78–Reference Grintal, Champeil-Potokar and Lavialle80), leading to a higher concentration of glutamate in the synapse cleft and prolongation of post- and presynaptic receptor activation(Reference Grintal, Champeil-Potokar and Lavialle80, Reference Robinson81). Such a mechanism could explain the increase of functional synapses observed in the present study after exposure to dietary DHA.
Our correlative analysis suggests that the CC started to rise when the DHA:AA threshold ratio was approximately 2:1. This observation suggests that it is the concurrent opposite variation of both PUFA, which underlies their effects on drebrin, syntaxin-3 and CC, although our data do not provide a clear mechanistic explanation. Quite interestingly, this DHA:AA threshold ratio coincided with the increase of syntaxin-3 levels and the translocation of drebrin from the cytosol to the membrane compartment, suggesting an involvement of these two proteins in regulating the size of the neuronal membrane surface. In agreement with our data, DHA administration was shown to increase the level of syntaxin-3 in the brain of rodents(Reference Wurtman, Cansev and Ulus76, Reference Chytrova, Ying and Gomez-Pinilla82), whereas the binding of DHA to syntaxin-3 induced its conformational change and complexing to SNAP-25 (synaptosomal-associated protein 25 kDa), thereby promoting membrane expansion(Reference Darios and Davletov28). Thus, up-regulation of syntaxin-3 is a plausible mechanism for the DHA-induced membrane expansion. Drebrin is another postsynaptic protein involved in dendritic spines scaffold(Reference Calon, Lim and Yang19, Reference Shim and Lubec64, Reference Sekino, Kojima and Shirao65) and required for optimal cognitive function(Reference Kojima, Hanamura and Yamazaki83). A loss of drebrin by antisense transfection reduced neurite outgrowth in neuroblastoma B104 cells(Reference Toda, Shirao and Uyemura84) and DHA treatment prevents the loss of drebrin in the Tg2576 model of Alzheimer's disease(Reference Calon, Lim and Yang19), a mouse line displaying neuronal atrophy(Reference Moolman, Vitolo and Vonsattel66). Overall, our data suggest that there is a threshold DHA:AA ratio in the brain leading to membrane expansion, in which the postsynaptic proteins, drebrin and syntaxin-3, play a role.
Given the crucial role of EC in cognition and memory, these differential effects of DHA and LNA on the basic electrophysiological properties of EC neurons could be instrumental to their action on CNS function. We have recently proposed that the enhancing effect of DHA on CC may reflect increased membrane surface area of neurons and improved capacity to communicate with other neurons and to collect/process a greater flow of brain information(Reference Arsenault, Julien and Tremblay30). The increased number of sEPSC detected in DHA-enriched neurons supports the idea of an enhanced integration into the neuronal network(Reference Arsenault, Julien and Tremblay30). Thus, these cellular effects of DHA may be directly related to some of the reported effects of DHA on memory/cognition function in animals(Reference Fedorova, Hussein and Baumann11, Reference Calon, Lim and Yang19, Reference Arsenault, Julien and Tremblay30, Reference Catalan, Moriguchi and Slotnick85, Reference Petursdottir, Farr and Morley86) and in humans(Reference Yurko-Mauro, McCarthy and Rom16, Reference Yurko-Mauro87). Accordingly, the membrane surface expansion induced by DHA may be therapeutic in neurodegenerative diseases involving neuronal atrophy, such as Alzheimer's disease(Reference Arsenault, Julien and Tremblay30), where the beneficial prophylactic effects of DHA have been reported in animal models(Reference Bousquet, Saint-Pierre and Julien8, Reference Calon and Cole13, Reference Calon, Lim and Yang19, Reference Petursdottir, Farr and Morley86) and suspected in human subjects(Reference Cunnane, Plourde and Pifferi15, Reference Cole and Frautschy88). The present data demonstrate the incapacity of dietary LNA to reproduce the effects of DHA on EC neuron physiology, and thus suggest that LNA may also be unable to exert the same beneficial effect on cognition as DHA, at least when consumed in an equimolar dose. Such contention is supported by a recent study showing that dietary LNA, when compared head-to-head with preformed DHA, does not achieve the same protective effect on sensorimotor gating, assessed by prepulse inhibition in C57Bl6 mice(Reference Fedorova, Alvheim and Hussein89). However, previous studies have shown that small increases in brain DHA produced by dietary LNA are enough to modify neurophysiological parameters such as synapse plasticity(Reference Lafourcade, Larrieu and Mato90), whereas the sequential administration of three doses of LNA increases neurogenesis and expression of key proteins involved in synaptic function (synaptophysin-1, vesicle-associated membrane protein 2 (VAMP-2), synaptosomal-associated protein 25 kDa (SNAP-25) and vesicular glutamate transporters 1 and 2 (VGLUT1 and VGLUT2))(Reference Blondeau, Nguemeni and Debruyne91). These two studies clearly show that dietary LNA can influence cerebral function, possibly through conversion into DHA or other n-3 PUFA.
Regulation of neuronal excitability by n-3 PUFA: network stability and anticonvulsive effect
LNA treatment did not fully reproduce another critical effect of DHA: the hyperpolarisation of the resting potential of EC neurons. Indeed, exposure to DHA during 10 months led to decrease of resting potential from − 60 to − 70 mV, whereas the effect of LNA was limited to a few mV. Our correlative analysis established a close link between the DHA:AA ratio and resting potential, suggesting that the limited capacity of LNA to modulate cerebral content of PUFA, compared to the DHA diet was again the most probable explanation. Since membrane hyperpolarisation is a basic mechanism of many anti-epileptic drugs, our results argue in favour of a potential therapeutic role of DHA, and not LNA, in the prevention of epileptic seizures. Anticonvulsant effects of DHA have been suggested by several authors(Reference Xiao and Li92–Reference Taha, Burnham and Auvin95) and a decrease in frequency and intensity of seizures following DHA intake have been reported in epileptic patients(Reference Schlanger, Shinitzky and Yam96). In summary, our data suggest that the change of ionic balance induced by dietary DHA can produce a superior anticonvulsant effect than LNA intake. However, it must be noted that very limited LNA-induced hyperpolarisation was apparently sufficient to reduce significantly the number of spontaneously active neurons. This surprising observation indicates that the consumption of LNA may nevertheless lead to enough brain DHA to reduce the production of non-specific activity of EC neurons within the network.
Nutritional implications
The study of the intrinsic properties provides basal information about neurons. They describe synaptic integration, the membrane surface, ion flux crossing the membrane and the intracellular ionic balance of each neuron. Thus, the present finding that equimolar dietary LNA cannot achieve the effects of DHA on intrinsic properties of EC neurons has important nutritional implications. Although people are becoming more educated concerning the potential benefit of n-3 PUFA, having to choose between LNA and EPA/DHA remains an important practical dilemma. Indeed, LNA and EPA/DHA are found in different foods and different nutritional supplements. Hence, consumption of LNA without an adequate supply of EPA/DHA may lead to sub-optimal brain DHA levels to change the intrinsic properties of neurons. Currently, the market is overwhelmed with products claiming to contain health-promoting n-3 PUFA, but containing only LNA without EPA or DHA. The present study provides one of the first direct pieces of evidence that the effect of a chronic dietary intake of preformed DHA on neuronal function cannot be replicated with LNA, despite comparable daily dosage. However, as human and rodent diets typically contain higher amounts of LNA and lower amounts of DHA than the animal diets used in the present study, it is possible that consumption of very high level of LNA could achieve the effect of preformed DHA by its conversion to DHA or other n-3 PUFA in the liver(Reference Brenna, Salem and Sinclair2, Reference Salem, Pawlosky and Wegher31–Reference Plourde and Cunnane33). Nevertheless, our results suggest that the beneficial effect of DHA on memory and cognition may be achieved more efficiently with dietary consumption of preformed DHA rather than with LNA.
Conclusion
We have shown that dietary LNA did not replicate the striking effect of DHA on EC neuronal function, such as changes in CC, resting potential and synaptic activity. Nevertheless, LNA supplementation significantly increased brain DHA levels and decreased AA levels, but not to the extent of direct intake of preformed DHA. The inability of LNA treatment to modulate passive properties of EC neurons may result from its inefficiency to raise the DHA:AA ratio above a threshold necessary to induce an increase of syntaxin-3 and the translocation of drebrin to the membrane compartment, as these two proteins have been involved in the regulation of membrane/cellular/neurite expansion. Since the basic properties of EC neurons modulated here by DHA are likely to be essential to higher brain function, our data support the preferential use of preformed DHA rather than LNA in CNS diseases.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (FC – MOP74443), the Alzheimer Society Canada (FC – ASC 0516) and the Canada Foundation for Innovation (10307). D. A. held a studentship from the Canadian Institutes of Health Research and the Fonds d'Enseignement et de Recherche of the Faculty of Pharmacy of Laval University. C. J. is supported by the Alzheimer Society Canada, the Fonds de la recherche en santé du Québec and the Fonds d'Enseignement et de Recherche of the Faculty of Pharmacy of Laval University. The work of F. C. was supported by a New Investigator Award from the Clinical Research Initiative and the Canadian Institutes of Health Research Institute of Aging (CAN-76833). Experimental design was made by F. C. and D. A. All of the electrophysiological and molecular experiments were performed by D. A. D. A. and C. J. performed the FA experiments. The management of the animal colony was under the supervision of C. J. The figures and analysis of the results were made by D. A., whereas the manuscript was written by D. A. and F. C. The authors declare that they have no actual or potential conflict of interest. The use of animals was approved by the Laval University Animal Ethics Committee in accordance with the standards of the Canadian Council on Animal Care.








