


INTRODUCTION
Cryptosporidium is a genus of apicomplexan parasites with species that infect all major vertebrate groups (Fayer, Reference Fayer2010; Ryan, Reference Ryan2010; Kváč et al. Reference Kváč, McEvoy, Stenger, Clark, Cacciò and Widmer2014). Infections can result in the diarrhoeal disease cryptosporidiosis, which can be chronic and even fatal in the absence of a competent immune response (Checkley et al. Reference Checkley, White, Jaganath, Arrowood, Chalmers, Chen, Fayer, Griffiths, Guerrant, Hedstrom, Huston, Kotloff, Kang, Mead, Miller, Petri, Priest, Roos, Striepen, Thompson, Ward, Van Voorhis, Xiao, Zhu and Houpt2015).
Early efforts to characterize Cryptosporidium – using descriptions of oocyst morphology, identification of surface antigens and isoenzyme analyses – lacked the resolution necessary to differentiate taxa infecting closely related hosts (Nichols et al. Reference Nichols, McLauchlin and Samuel1991; Nina et al. Reference Nina, McDonald, Deer, Wright, Dyson, Chiodini and McAdam1992; Ogunkolade et al. Reference Ogunkolade, Robinson, McDonald, Webster and Evans1993; McLauchlin et al. Reference McLauchlin, Casemore, Moran and Patel1998). Molecular tools have revealed tremendous genetic diversity in the genus Cryptosporidium, and more than 30 species and tens of genotypes have been described to date (Ryan et al. Reference Ryan, Fayer and Xiao2014; Holubová et al. Reference Holubová, Sak, Horčičková, Hlásková, Květoňová, Menchaca, McEvoy and Kváč2016; Ježková et al. Reference Ježková, Horčičková, Hlásková, Sak, Květoňová, Novák, Hofmannová, McEvoy and Kváč2016; Kváč et al. Reference Kváč, Havrdová, Hlásková, Daňková, Kanděra, Ježková, Vítovec, Sak, Ortega, Xiao, Modrý, Chelladurai, Prantlová and McEvoy2016). One hypothesis holds that Cryptosporidium diversification is promoted by coevolutionary interactions with hosts, and this is supported by the findings that some closely related Cryptosporidium spp. infect a narrow range of closely related hosts. However, other species can infect a broad range of distantly related hosts, suggesting that coevolution is not the only driver of Cryptosporidium diversification.
Rodents are a useful model to study Cryptosporidium diversification. These ubiquitous mammals comprise about 40% of the mammalian diversity, with over 2200 species in 31 families and 481 genera, occupy a wide range of habitats, are extremely fecund and host diverse Cryptosporidium species and genotypes (Kváč et al. Reference Kváč, McEvoy, Stenger, Clark, Cacciò and Widmer2014). In addition to hosting species with a broad host specificity, including Cryptosporidium muris, Cryptosporidium parvum, and Cryptosporidium ubiquitum, rodents host more than 20 Cryptosporidium genotypes that appear to have a relatively narrow host range. For example, rats are commonly infected with Cryptosporidium rat genotypes I–IV, which have not been detected in other rodent species (Kimura et al. Reference Kimura, Edagawa, Okada, Takimoto, Yonesho and Karanis2007; Paparini et al. Reference Paparini, Jackson, Ward, Young and Ryan2012; Ng-Hublin et al. Reference Ng-Hublin, Singleton and Ryan2013; Zhao et al. Reference Zhao, Wang, Zhao, Qi, Zhao, Zhang, Li and Liu2015). Similarly, different species/genotypes of Cryptosporidium infect the squirrel tribes Marmotini and Sciurini (Stenger et al. Reference Stenger, Clark, Kváč, Khan, Giddings, Prediger and McEvoy2015b ). Narrowly specific Cryptosporidium species/genotypes may diverge as a consequence of host divergence, as was observed in the house mouse, where two subspecies (Mus musculus musculus and M. m. domesticus) that diverged 0·5 Mya (Bonhomme and Searle, Reference Bonhomme, Searle, Macholán, Baird, Munclinger and Piálek2012) hosted different subtypes of C. tyzzeri (Kváč et al. Reference Kváč, McEvoy, Loudová, Stenger, Sak, Květoňová, Ditrich, Rašková, Moriarty, Rost, Macholán and Piálek2013).
The Cricetidae, at almost 600 species, is the second-largest family of mammals, comprising the subfamilies Cricetinae (hamsters), Sigmodontinae (including the cotton rat, climbing mice and water mice), Tylomyinae (including vesper rats and climbing rats), Neotominae (including deer mice and woodrats) and Arvicolinae (voles, muskrats and lemmings). The Cricetinae are exclusively Palearctic, being found in central and eastern Europe and parts of Asia. The Neotominae, Thylomyinae and Sigmodontinae are Nearctic/Neotropical, and are predominantly found in North, Central and South America, respectively. The Holarctic Arvicolinae underwent an explosive radiation, resulting in 151 extant species in 28 genera, as they dispersed from Asia to Europe and North America (NA) (Steppan et al. Reference Steppan, Adkins and Anderson2004; Wilson and Reeder, Reference Wilson and Reeder2005).
Several Cryptosporidium genotypes appear to be specific to cricetids, and some may be specific for cricetid subfamilies. Cryptosporidium vole genotype and muskrat genotypes I and II have been reported only in arvicolines (voles and muskrats). Similarly, Cryptosporidium deer mouse genotypes I-IV appear mostly restricted to deer mice, in the subfamily Neotominae (Perz and Le Blancq, Reference Perz and Le Blancq2001; Xiao et al. Reference Xiao, Sulaiman, Ryan, Zhou, Atwill, Tischler, Zhang, Fayer and Lal2002; Zhou et al. Reference Zhou, Fayer, Trout, Ryan, Schaefer and Xiao2004; Feng et al. Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Ziegler et al. Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007; Lv et al. Reference Lv, Zhang, Wang, Jian, Zhang, Ning, Wang, Feng, Wang, Ren, Qi and Xiao2009; Robinson et al. Reference Robinson, Chalmers, Stapleton, Palmer, Watkins, Francis and Kay2011; Ruecker et al. Reference Ruecker, Matsune, Wilkes, Lapen, Topp, Edge, Sensen, Xiao and Neumann2012).
Here we report a study on Cryptosporidium infecting wild cricetid rodent populations in NA (at sites in North Dakota, Minnesota, South Dakota and Tennessee) and Europe (at sites in the Czech Republic and Slovakia). Data from the study contribute to the understanding of Cryptosporidium evolution in closely related hosts on different continents.
MATERIALS AND METHODS
Ethics statement
The research was conducted under ethical protocols approved by the Institute of Parasitology, Biology Centre and Central Commission for Animal Welfare, Czech Republic (protocol nos. 071/2010 and 114/2013) and North Dakota State University Institutional Animal Care and Use Committee (protocol A11060).
Sample collection – NA
Meadow voles (Microtus pennsylvanicus), southern red-backed voles (Myodes gapperi), muskrats (Ondatra zibethicus) and Peromyscus mice (deer mice, Peromyscus maniculatus and white-footed mice, Peromyscus leucopus, were not distinguished in this study) were sampled in North Dakota, South Dakota and Minnesota. Woodland voles (Microtus pinetorum) and Peromyscus mice were sampled in an area Tennessee. Except for muskrats, North American cricetids were live captured in Sherman box traps and fecal samples were collected from the trap or directly from the animal during handling. Captured animals were ear-tagged and released. Animals that died in traps were dissected and samples of intestinal contents were examined. Muskrats were sampled by collecting feces from muskrat mounds. All samples were stored at 4 °C prior to DNA extraction.
Sample collection – Europe (EU)
Common voles (Microtus arvalis) and bank voles (Myodes glareolus) were captured in Sherman box traps in the Czech Republic and Slovakia. Trapped animals were euthanized and samples were collected from the intestines following dissection.
Polymerase chain reaction amplification and sequencing
For North American samples, DNA was isolated from samples by alkaline digestion, phenol-chloroform extraction and purified using a QIAmp DNA Stool Mini Kit (Qiagen, Valencia, CA) as previously described (Peng et al. Reference Peng, Wilson, Holland, Meshnick, Lal and Xiao2003; Feltus et al. Reference Feltus, Giddings, Schneck, Monson, Warshauer and McEvoy2006). For European samples, 200 mg of feces was homogenized by bead disruption using FastPrep-24 (Biospec Products, Bartlesville, OK) for 60 s at a speed 5·5 m/s. Total DNA was extracted using the PSP Spin Stool DNA Kit (Invitek, Berlin, Germany).
DNA was stored at −20 °C until used in PCR assays. Fragments of the Cryptosporidium small subunit (SSU) and actin genes were amplified using nested PCR assays as described previously (Xiao et al. Reference Xiao, Singh, Limor, Graczyk, Gradus and Lal2001; Sulaiman et al. Reference Sulaiman, Lal and Xiao2002). Secondary products were visualized with SYBR Green or ethidium bromide following electrophoresis on an agarose gel.
PCR products were purified (Wizard SV, Promega, Madison, WI or GenElute™ Gel Extraction Kit, Sigma-Aldrich, St. Louis, MO) and sequenced in both directions with secondary primers using a BigDye Terminator v3·1 cycle sequencing kit in an ABI Prism 3130 genetic analyzer (Applied Biosystems, Carlsbad, CA). Sequences were assembled using SeqMan (DNAStar, Madison, WI).
Phylogenetic analysis
Sequences were aligned using the MAFFT version 7 online server with automatic selection of alignment strategy (http://mafft.cbrc.jp/alignment/server/) (Katoh and Standley, Reference Katoh and Standley2013). Alignments were manually edited and phylogenetic analyses were performed using MEGA 6·0 (Tamura et al. Reference Tamura, Stecher, Peterson, Filipski and Kumar2013). The evolutionary history of aligned sequences was inferred using the maximum likelihood (ML) method (Saitou and Nei, Reference Saitou and Nei1987), with the substitution model that best fit the alignment selected using the Bayesian information criterion. The Hasegawa–Kishino–Yano model (Hasegawa et al. Reference Hasegawa, Kishino and Yano1985) was selected for SSU alignments, and the general time reversible model (Tavaré, Reference Tavaré and Miura1986) was selected for actin and concatenated actin-SSU alignments. Both models were used under an assumption that rate variation among sites was gamma distributed. A bootstrap consensus tree was inferred from 1000 pseudoreplicates. Phylogenetic analyses, including analysis of substitution model goodness of fit, were carried out using MEGA 6·0. Phylogenetic trees were edited for style using Adobe Illustrator CS5·1 (AdobeSystems, Inc., San Jose, CA).
Principal coordinate analysis
Sequences were aligned with ClustalW (Thompson et al. Reference Thompson, Higgins and Gibson1994) and manually trimmed to remove terminal nucleotides not present in all sequences. For each alignment (SSU, actin, and concatenated SSU-actin sequences), a matrix of pairwise distances between sequences was constructed using the program dist.seqs in mothur (Schloss et al. Reference Schloss, Westcott, Ryabin, Hall, Hartmann, Hollister, Lesniewski, Oakley, Parks, Robinson, Sahl, Stres, Thallinger, Van Horn and Weber2009). Distance matrices were imported into GenAlEx (Peakall and Smouse, Reference Peakall and Smouse2012) and distances visualized by Principal Coordinate analysis (PCoA).
Statistical analysis
Prevalence was calculated by dividing the number of positive individuals by the total number of individuals sampled. Differences in Cryptosporidium prevalence were determined by Chi-square analysis using a 5% significance level. Analyses were performed using the statistical program R (R Core, 2013). The statistical significance of clusters visualized by PCoA was tested using ANOSIM in mothur (Clarke, Reference Clarke1993).
RESULTS
In total 1089 animals from the family Cricetidae were sampled at locations in NA (596 animals) and Europe (493 animals). A total of 681 samples were obtained from the 596 North American cricetids. The greater number of samples than animals was due to some animals from NA being sampled multiple times. All animals from Europe were sampled only once. Overall, 33·2% (362/1089) of cricetids tested positive for Cryptosporidium, with a greater prevalence in cricetids from NA (50·7%; 302/596) than Europe (12·1%; 60/493). Excluding repeat samples from the same animal, the prevalence in North American cricetids was 48·7% (290/596). In NA, the lowest prevalence was in muskrats (9·5%; 4/42) (P < 0·05). Peromyscus mice (56·6%; 99/175), southern red-backed voles (55·6%; 15/27), meadow voles (52·4%; 163/311) and woodland voles (51·2%; 21/41) had a similar prevalence. In Europe, the prevalence in common voles and bank voles was 14·2% (50/353) and 7·1% (10/140), respectively (P < 0·05).
Analysis of SSU sequences
Cryptosporidium SSU sequences were obtained from 126 animals and relationships among sequences were examined using PCoA and ML analysis (online Supplementary Fig. S1).
We used PCoA to visualize the matrix of pairwise genetic distances in a simplified, two-dimensional Euclidean space. Sixty-three percent of the SSU sequence variation was explained by two principal Coordinate, along which sequences separated into three groups that were statistically different from each other (G1–G3) (online Supplementary Fig. S1). These PCoA groups were overlaid on a ML tree constructed from Cryptosporidium SSU sequences (online Supplementary Fig. S1).
G1 included 97 sequences from all hosts and geographic locations examined in the study. Within G1, sequences from 28 meadow voles, 20 common voles, a muskrat and a Peromyscus mouse clustered with muskrat genotype II in the ML tree. G1 also included sequences clustering with C. ubiquitum, deer mouse genotypes I–IV, W29 genotype, fox genotype, vole genotype, chipmunk genotype IV and sequences that did not cluster with previously described species or genotypes.
Sequences from G2 formed a reasonably well-supported clade in the ML tree, within which sequences from meadow voles in NA and common voles in Europe formed separate clusters. This clade also included Cryptosporidium W12 genotype (AY007254), which was previously isolated from surface water in New York but has not been reported previously in an animal host. None of the sequences in the present study shared 100% identity with the W12 genotype.
Nested within a well-supported clade that included all sequences from G3, sequences from meadow voles and a muskrat in NA formed a sister group with sequences from common voles in Europe. The North American group included sequences previously identified as muskrat genotype I. A third group in this clade comprised sequences from bank voles in Europe, a sequence previously isolated from a yellow-necked mouse (Apodemus flavicollis) in Sweden (JN172968), and a sequence isolated from water in the UK (HM015876).
In some cases, divergent SSU gene sequences were obtained from different samples of the same animal. Sequences from three samples of the same Peromyscus mouse (1835-Pero-NA, 1851-Pero-NA, and 1852-Pero-NA) shared between 99·1 and 99·6% identity with each other and clustered with deer mouse genotype IV, which was previously isolated from a Peromyscus mouse in New York (EF641019). The samples were collected on 2 consecutive days: 1835-Pero-NA was obtained from the feces of the animal on the first day. The animal was released and was recaptured the next day, at which point the animal died in the trap, was dissected and 1851-Pero-NA and 1852-Pero-NA were obtained from the intestine. A fourth sequence (1848-Pero-NA) from the same animal, which was isolated from feces on the second day, clustered with the W29 genotype (JQ413356) as a sister group to deer mouse genotype IV, sharing between 98·1% and 98·5% sequence identity with 1835-Pero-NA, 1851-Pero-NA and 1852-Pero-NA.
Analysis of actin and concatenated actin-SSU gene sequences
Actin sequences were obtained from 70 samples and relationships among sequences were determined by PCoA and ML analysis. Sequences separated into five statistically different groups in the PCoA (G1-G5), and these groups were highlighted on the ML tree (Fig. 1 and online Supplementary Fig. S2).
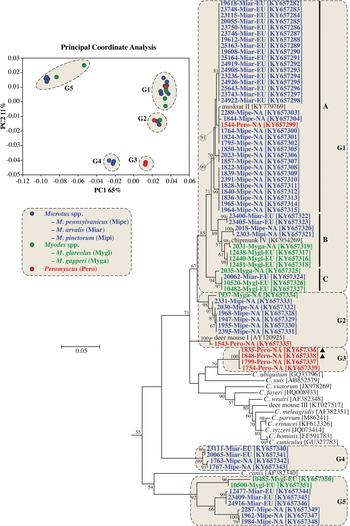
Fig. 1. Principle Coordinate Analysis (PCoA) and a maximum likelihood (ML) tree based on actin gene sequences. The five major PCoA groups (G1-G5) are highlighted against a cream background with dashed border on the ML tree. G1 is further broken down into three subgroups (A–C). Sequences from this study are identified by region (NA for NA and EU for Europe), and they are colour coded based on the genus of the host from which the sample was obtained (blue for Microtus spp., green for Myodes spp., and red for Peromyscus spp.). A solid black triangle (▲) identifies isolates from the same animal. The ML tree was rooted with an actin sequence from Plasmodium falciparum (accession number: EF472536). Due to limited space, the outgroup and some basal Cryptosporidium taxa are not shown. An expanded tree is shown in online Supplementary Fig. S2.
Sequences in G1 formed three major clades in the ML tree (labelled A–C in Fig. 1 and online Supplementary Fig. S2). Clade A, which had 71% bootstrap support, comprised four closely-related subclades. One of the subclades comprised entirely of sequences from bank voles in Europe. Two subclades included sequences from North American meadow voles only, and one subclade contained sequences from five meadow voles and a Peromyscus mouse in this study and a sequence previously identified as muskrat genotype II. Clade B had 89% bootstrap support and included four subclades, two of which formed closely related sister groups. One of the sister groups included a sequence from a North American red-backed vole (2031-Myga-NA) and a sequence previously isolated from a North American eastern chipmunk. The other sister group comprised three identical sequences from bank voles in the Czech Republic. A third subclade comprised sequences from a meadow vole and woodland vole in NA. A fourth subclade included sequences from the common vole in Europe. Clade C, which had 94% bootstrap support, included identical sequences from a common vole and two bank voles in Europe, and a sequence from a red-backed vole in NA that clustered separately, sharing 99·0% identity with the sequences from European voles.
Sequences in G2 formed two clades. One of the clades included sequences from meadow voles that were identified as the vole genotype in the SSU phylogeny, a sequence from a woodland vole (2331-Mipi-NA) and a sequence from a red-backed vole (1937-Myga-NA). A second clade in G2 contained 1543-Pero-NA from a Peromyscus mouse and a sequence previously identified as deer mouse genotype II; this clade was more closely related to sequences from Peromyscus mice in G3 than sequences from voles in G2. The four sequences from Peromyscus mice in G3 included 1835-Pero-NA and 1848-Pero-NA, which were from a single animal and were identified as deer mouse genotype IV and W29 genotype, respectively, at the SSU locus (online Supplementary Fig. S1). G4 and G5 formed well-supported clades in the ML tree, and nested within each were sequences that clustered by host/geographic location.
PCoA and ML analysis of SSU and actin gene sequences in concatenation produced similar groupings to actin sequences. The exception was 1543-Pero-NA1, which was not part of a PCoA group in the analysis of concatenated sequences (Fig. 2 and online Supplementary Fig. S3).
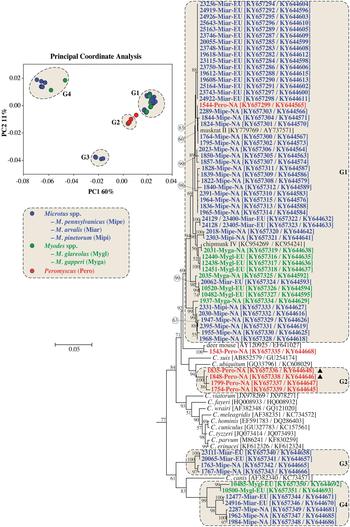
Fig. 2. Principle Coordinate Analysis (PCoA) and a maximum likelihood (ML) tree based on concatenated actin and small subunit rRNA (SSU) gene sequences. The four major PCoA groups (G1–G4) are highlighted against a cream background with dashed border on the ML tree. Sequences from this study are identified by region (NA for NA and EU for Europe), and they are colour coded based on the genus of the host from which the sample was obtained (blue for Microtus spp., green for Myodes spp. and red for Peromyscus spp.). A solid black triangle (▲) identifies isolates from the same animal. The ML tree was rooted with a concatenated actin/SSU sequence from Plasmodium falciparum (accession numbers: EF472536/JQ627149). Due to limited space, the outgroup and some basal Cryptosporidium taxa are not shown. An expanded tree is shown in online Supplementary Fig. S3.
DISCUSSION
Cryptosporidium diversity may result, in part, from a close association with diverging host species. This model of evolution is supported by evidence that Cryptosporidium has diverged with subspecies of the house mouse, Mus musculus (Kváč et al. Reference Kváč, McEvoy, Loudová, Stenger, Sak, Květoňová, Ditrich, Rašková, Moriarty, Rost, Macholán and Piálek2013). Two subspecies, Mus musculus musculus and M. m. domesticus, which diverged after becoming geographically isolated about 0·5 Mya, host genetically and biologically distinct subtypes of C. tyzzeri, and the subtypes have remained host-specific despite the establishment of secondary contact between M. m. musculus and M. m. domesticus. The study by Kváč et al. (Reference Kváč, McEvoy, Loudová, Stenger, Sak, Květoňová, Ditrich, Rašková, Moriarty, Rost, Macholán and Piálek2013) demonstrated that knowledge of the timing of host divergence can be used to understand the dynamics of parasite divergence. Using a similar approach in the present study, we examined Cryptosporidium diversity in rodent species from the family Cricetidae.
Cryptosporidium from voles exhibited considerable SSU sequence heterogeneity, which is consistent with previous studies on Cryptosporidium from voles and muskrats. Most sequences clustered with previously named Cryptosporidium genotypes, including muskrat genotype I, muskrat genotype II, vole genotype and fox genotype. Sequences clustering with muskrat genotypes I and II were rarely detected in hosts other than voles, which is consistent with previous reports that these genotypes primarily infect voles, and are found less frequently in muskrats, Peromyscus mice and foxes (Zhou et al. Reference Zhou, Fayer, Trout, Ryan, Schaefer and Xiao2004; Feng et al. Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Ziegler et al. Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007; Robinson et al. Reference Robinson, Chalmers, Stapleton, Palmer, Watkins, Francis and Kay2011; Ruecker et al. Reference Ruecker, Matsune, Wilkes, Lapen, Topp, Edge, Sensen, Xiao and Neumann2012). Therefore, despite the assigned genotype names, voles should be considered the major host for muskrat genotypes I and II. Similarly, we found that sequences clustering with the W12 and vole genotypes were exclusive to voles. The vole genotype has been identified previously in meadow voles (Feng et al. Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Ziegler et al. Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007), but this is the first report of a host for the W12 genotype, which was previously reported only in water (Feng et al. Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Ruecker et al. Reference Ruecker, Braithwaite, Topp, Edge, Lapen, Wilkes, Robertson, Medeiros, Sensen and Neumann2007).
The 102 variants detected among 134 SSU sequences examined suggests that cricetids host diverse Cryptosporidium taxa. The multiple-taxa hypothesis is predicated on the assumption that SSU sequences are orthologous, which is generally true; however, SSU sequences could also have a paralogous relationship. Some apicomplexans, including Cryptosporidium, can have divergent SSU paralogues that complicate the accurate reconstruction of evolutionary histories (Le Blancq et al. Reference Le Blancq, Khramtsov, Zamani, Upton and Wu1997; Xiao et al. Reference Xiao, Limor, Li, Morgan, Thompson and Lal1999; Morgan et al. Reference Morgan, Monis, Xiao, Limor, Sulaiman, Raidal, O'Donoghue, Gasser, Murray, Fayer, Blagburn, Lal and Thompson2001; Kimura et al. Reference Kimura, Edagawa, Okada, Takimoto, Yonesho and Karanis2007; Santín and Fayer, Reference Santín and Fayer2007; Lv et al. Reference Lv, Zhang, Wang, Jian, Zhang, Ning, Wang, Feng, Wang, Ren, Qi and Xiao2009; Sevá Ada et al. Reference Sevá Ada, Funada, Richtzenhain, Guimarães, Souza, Allegretti, Sinhorini, Duarte and Soares2011; Ikarashi et al. Reference Ikarashi, Fukuda, Honma, Kasai, Kaneta and Nakai2013; Ng-Hublin et al. Reference Ng-Hublin, Singleton and Ryan2013; Stenger et al. Reference Stenger, Clark, Kváč, Khan, Giddings, Dyer, Schultz and McEvoy2015a ). Ideally, paralogy should be tested in a single lineage, where it can be confirmed that the divergent SSU sequences are present in the same genome (Le Blancq et al. Reference Le Blancq, Khramtsov, Zamani, Upton and Wu1997). This is rarely possible in field studies on Cryptosporidium in complex fecal samples due to a lack of tools to propagate individual strains. Paralogy should be suspected when divergent SSU sequences co-occur in samples without the divergence of other polymorphic loci, such as actin and HSP70 (Stenger et al. Reference Stenger, Clark, Kváč, Khan, Giddings, Dyer, Schultz and McEvoy2015a ). A limitation of this approach is the possibility that comparatively rare SSU and actin/HSP70 polymorphisms may not be detected by direct sequencing of PCR amplicons. In the present study, three isolates clustered with deer mouse genotype IV and three isolates clustered with the closely related W29 genotype at the SSU locus. All isolates clustering with deer mouse genotype IV and one of the W29 isolates were from a single animal and had identical sequences at the actin locus. Therefore, deer mouse genotype IV and W29 genotype could represent SSU paralogues rather than closely related taxa. Feng et al. (Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007) similarly suggested that deer mouse genotypes I and II, which were detected in a single deer mouse, may be paralogues. Because paralogy is difficult to confirm in Cryptosporidium, when it is suspected, genes other than SSU should be used for phylogenetic reconstructions.
We found that, with few exceptions, the cricetid subfamilies Neotominae (Peromyscus mice) and Arvicolinae (voles and muskrats), which diverged about 19 Mya (Steppan et al. Reference Steppan, Adkins and Anderson2004), hosted phylogenetically distinct Cryptosporidium species and genotypes. Deer mouse genotypes I–IV, W29 genotype and C. ubiquitum were exclusively found in Peromyscus mice. Cryptosporidium ubiquitum, which was found in a single Peromyscus mouse, has a broad host specificity that includes many rodent and non-rodent mammals. We previously detected C. ubiquitum and deer mouse genotype III in squirrels from the same area as the Peromyscus mice sampled in the present study (Stenger et al. Reference Stenger, Clark, Kváč, Khan, Giddings, Prediger and McEvoy2015b ). Feng et al. (Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007) also found C. ubiquitum and deer mouse genotype III in Peromyscus mice and squirrels in the eastern USA, suggesting frequent transmission between these different rodent families. This could be explained by the propensity of Peromyscus mice and squirrels to occupy the same habitat (Brunner et al. Reference Brunner, Duerr, Keesing, Killilea, Vuong and Ostfeld2013). In contrast, voles and Peromyscus mice are known to spatially segregate within grassland habitats, limiting interspecific interactions (Bowker and Pearson, Reference Bowker and Pearson1975).
Cryptosporidium genotypes infecting Microtus spp. and Myodes spp. generally clustered separately in actin and actin-SSU phylogenies, regardless of geographic location, suggesting that Cryptosporidium has coevolved with these cricetid genera. This is consistent with the Myodes-Microtus divergence time estimate of 5·76–9 Mya (Robinson et al. Reference Robinson, Catzeflis, Briolay and Mouchiroud1997; Conroy and Cook, Reference Conroy and Cook1999), before they colonized NA. Myodes likely colonized NA from Eurasia in the late Pliocene (3·6–2·58 Mya) to early Pleistocene (2·58–0·78 Mya) (Cook et al. Reference Cook, Runck and Conroy2004) and Microtus followed sometime later (Martin, Reference Martin2003).
Although this study found that cricetids are frequently infected with Cryptosporidium, the species/genotypes pose little threat to human health. Only C. ubiquitum, which we detected in a single Peromyscus mouse, has been associated with human disease (Chalmers et al. Reference Chalmers, Smith, Elwin, Clifton-Hadley and Giles2011; Cieloszyk et al. Reference Cieloszyk, Goñi, García, Remacha, Sánchez and Clavel2012; Li et al. Reference Li, Xiao, Alderisio, Elwin, Cebelinski, Chalmers, Santín, Fayer, Kváč, Ryan, Sak, Stanko, Guo, Wang, Zhang, Cai, Roellig and Feng2014).
In summary, North American and European cricetids host diverse Cryptosporidium spp., which in many cases appear to have coevolved with their hosts. Using only sequences of SSU to infer evolutionary relationships of Cryptosporidium may lead to erroneous conclusions, so it is recommended to use other polymorphic loci in phylogenetic analyses.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182017001524
FINANCIAL SUPPORT
The authors gratefully acknowledge funding support from the US Department of Agriculture National Research Initiative (Project # 2008-35102-19260), the National Institute of Allergy and Infectious Diseases (1R15AI122152-01A1), the Czech Science Foundation (15-01090S), the Grant Agency of University of South Bohemia (002/2016/Z and 098/2016/Z), and the North Dakota Water Resources Research Institute, North Dakota State University Graduate School.



