
Contents
Research Article
The Background in X-Ray Powder Diffractograms: A Case Study of Rietveld Analysis of Minor Phases Using Ni-Filtered and Graphite-Monochromated Radiation
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 63-70
-
- Article
- Export citation
The Determination of Low Levels of Quartz in Commercial Kaolins by X-ray Diffraction
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 71-76
-
- Article
- Export citation
A Simplified Correction for Surface Roughness Effects in X-Ray Diffractometric Investigations
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 77-82
-
- Article
- Export citation
Notes on Slits and Monochromators in Accurate Powder Diffractometry
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 83-88
-
- Article
- Export citation
Intensity Calibration Curves for Bragg-Brentano X-Ray Diffractometers
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 89-94
-
- Article
- Export citation
Validation of Reference Patterns by Search/Matching
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 95
-
- Article
- Export citation
A Review of the XRD Data of the Phases Present in the CaO-SrO-PbO System
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 96-98
-
- Article
- Export citation
The Crystal Structure of Pressure-Induced Phases of In2Te3and Ga2Te3
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 99-102
-
- Article
- Export citation
The Crystal Structure of K2REZr(PO4)3(RE = Y, Gd) Isotypic with Langbeinite
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 103-106
-
- Article
- Export citation
Powder Crystal Data for the High-Temperature Phases Cu4In, Cu9In4(h) and Cu2In(h)
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 107-108
-
- Article
- Export citation
Powder X-Ray Diffraction Data for Ca2Bi2O5and C4Bi6O13
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 109-111
-
- Article
- Export citation
-
Single crystals and powder samples of Ca2Bi5O5and Ca4Bi6O13have been synthesized and studied using single crystal X-ray diffraction as well as X-ray and neutron powder diffraction. Unit cell dimensions were calculated using a least squares analysis that refined to a δ2θof no more than 0.03°. A triclinic cell was found with space group
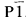 , a = 10.1222(7), b = 10.1466(6), c = 10.4833(7) Å. α= 116.912(5), β= 107.135(6) and γ= 92.939(6)°, Z = 6 for the Ca2Bi2O5compound. An orthorhombic cell was found with space group C2mm, a = 17.3795(5), b = 5.9419(2) and c = 7.2306(2) Å, Z = 2 for the Ca4Bi6O13compound.
, a = 10.1222(7), b = 10.1466(6), c = 10.4833(7) Å. α= 116.912(5), β= 107.135(6) and γ= 92.939(6)°, Z = 6 for the Ca2Bi2O5compound. An orthorhombic cell was found with space group C2mm, a = 17.3795(5), b = 5.9419(2) and c = 7.2306(2) Å, Z = 2 for the Ca4Bi6O13compound.
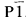 , a = 10.1222(7), b = 10.1466(6), c = 10.4833(7) Å.
, a = 10.1222(7), b = 10.1466(6), c = 10.4833(7) Å. Improved X-Ray Powder Diffraction Data for Franckeite
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 112-114
-
- Article
- Export citation
Powder X-Ray Diffraction Data for the Superconducting Phase TlCaBa2Cu2O7−δ
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 115-116
-
- Article
- Export citation
Errata
X-Ray Powder Diffraction Data of a New Compound Na2Ca4Mg2Si4O15
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 117-118
-
- Article
-
- You have access
- Export citation
International Report
Calendar of Meetings
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 117-120
-
- Article
-
- You have access
- Export citation
Meeting Reports
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 120-121
-
- Article
- Export citation
Short Courses and Workshops
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 122
-
- Article
-
- You have access
- Export citation
Commercial Announcements
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 123
-
- Article
-
- You have access
- Export citation
Front matter
PDJ volume 7 issue 2 Cover and Front matter
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f19
-
- Article
-
- You have access
- Export citation
Back matter
PDJ volume 7 issue 2 Cover and Back matter
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b4
-
- Article
-
- You have access
- Export citation