
Contents
Research Article
X-Ray Powder Diffraction Characterization of BaR2CuO5 (R=Yttrium and the Lanthanides) and Related Compounds
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-8
-
- Article
- Export citation
Determination of Amorphous Phase in Quartz Powder by X-Ray Powder Diffractometry
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 9-13
-
- Article
- Export citation
Sodium Copper Oxalate Dihydrate: Na2Cu (C2O4)2. 2H2O Synthesis, Characterization, Morphology and Optical Properties
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 14-18
-
- Article
- Export citation
Unit-Cell Dimensions of Two-Dimensionai Clay Minerals
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 19-20
-
- Article
- Export citation
X-Ray Powder Data for Two Tetracyclines: 6-Methylene-5-Hydroxytetracycline Hydrochloride (Methacycline Hydrochloride), C22H23N2O8Cl, and 6-Demethyl-7-Chlorotetracycline Hydrochloride Trihydrate (Declomycin Hydrochloride), C21H28O11N2Cl2
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 21-23
-
- Article
- Export citation
The Crystal Structure of BaY2O4, Isotypic with SrY2O4
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 24-25
-
- Article
- Export citation
High Quality Powder Diffraction Data from an Impure Specimen: Triphylite, Li(Fe,Mn)PO4
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 26-28
-
- Article
- Export citation
Revised Crystal Data of Zr(MoO4)2, L.T. Form
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 29-30
-
- Article
- Export citation
Crystal Data for Thirteen Tellurates MIBaNbTe2O9 (MI = Li,Na,K,Rb,Ag) and
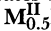 BaNbTe2O9 (MII = Mg,Ca,Sr,Ba,Cu,Zn,Cd,Pb)
BaNbTe2O9 (MII = Mg,Ca,Sr,Ba,Cu,Zn,Cd,Pb)
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 31-33
-
- Article
- Export citation
-
Thirteen isotypic tellurates having the stoichiometries MIBaNbTe2O9 and
BaNbTe2O9, with MI = Li,Na,K,Rb,Ag and MII = Mg,Ca,Sr,Ba,Cu,Zn,Cd,Pb, have been synthesized by solid state reaction. Single crystals of KBaNbTe2O9 were obtained. The compounds crystallize in the orthorhombic space group P212121 [19]. Unit-cell parameters for the 13 compounds and powder diffraction patterns for three representative compounds, KBaNbTe2O9, Ba1.5NbTe2O9 and Cd0.5BaNbTe2O9, are given.
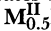 BaNbTe
BaNbTeX-Ray Powder Diffraction Data for Wolfeite: (Fe0.59Mn0.40Mg0.01)2PO4(OH)
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 34-35
-
- Article
- Export citation
Powder Diffraction Data for Synthetic Pargasite, Scandian Pargasite and Their Fluorine Analogues: NaCa2Mg4(Al,Sc)Si6Al2O22(OH,F)2
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 36-39
-
- Article
- Export citation
Standard X-Ray Diffraction Powder Patterns of Fourteen Ceramic Phases
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 40-54
-
- Article
- Export citation
Departments
General Announcements
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 55
-
- Article
-
- You have access
- Export citation
Calendar of Meetings
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 56-57
-
- Article
-
- You have access
- Export citation
Correspondents Reports
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 57-58
-
- Article
- Export citation
JCPDS-ICDD Subcommittee Activities
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 59-60
-
- Article
- Export citation
Short Courses and Workshops
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 61
-
- Article
-
- You have access
- Export citation
Computer Comments
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 62-63
-
- Article
- Export citation
Commercial Announcements
-
- Published online by Cambridge University Press:
- 10 January 2013, p. 64
-
- Article
-
- You have access
- Export citation
Front matter
PDJ volume 4 issue 1 Cover and Front matter
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f19
-
- Article
-
- You have access
- Export citation