


INTRODUCTION
Parasite assemblage structure can change through local colonization and extinction of individual lineages, which depend on several attributes of hosts (Poulin, Reference Poulin1997). For example, the geographic range of the host has been shown to positively influence parasite richness: host species with larger ranges would presumably be exposed to, and colonized by, more parasite species over evolutionary time (Poulin, Reference Poulin1997, Reference Poulin2007). Indeed, this pattern has been found among several types of parasites and assemblages in different host taxa (Poulin, Reference Poulin1997, Reference Poulin2007; Kamiya et al. Reference Kamiya, O'Dwyer, Nakagawa and Poulin2014). The effects of ecological and evolutionary processes on parasite assemblages can be revealed, to some degree, by comparing parasite assemblages among host species and/or among localities, and correlating variation in assemblage similarity with ecological and evolutionary factors. Comparing assemblages across host species, the extent of geographic overlap and phylogenetic relatedness of hosts may influence parasite assemblages. Davies and Pedersen (Reference Davies and Pedersen2008) found evidence for both these factors in the greater similarity of pathogen assemblages between more closely related primates and between primates sharing more of their geographic ranges. In multi-host vector-borne pathogens, comparisons of parasite assemblages across localities are also worthwhile, as turnover may result from, for example, climate and habitat differences (which are most likely to affect parasites through their ectothermic, moisture-dependent vectors), and host assemblage turnover (e.g. Ishtiaq et al. Reference Ishtiaq, Clegg, Phillimore, Black, Owens and Sheldon2010; Svensson-Coelho and Ricklefs, Reference Svensson-Coelho and Ricklefs2011). Finally, assemblage similarity of free-living organisms is expected to decrease with increasing geographic distance (Nekola and White, Reference Nekola and White1999; Soininen et al. Reference Soininen, McDonald and Hillebrand2007), which has also been demonstrated in some assemblages of parasitic organisms (Poulin, Reference Poulin2003; Krasnov et al. Reference Krasnov, Shenbrot, Mouillot, Khokhlova and Poulin2005; Svensson-Coelho and Ricklefs, Reference Svensson-Coelho and Ricklefs2011).
Haemosporida of the genera Plasmodium and Haemoproteus are vector-borne protozoan parasites that complete asexual reproduction in their avian hosts (Valkiūnas, Reference Valkiūnas2005). These parasites are nearly worldwide in distribution, abundant and diverse in most bird orders and families, and infect a wide range of invertebrate dipteran vectors (Valkiūnas, Reference Valkiūnas2005; Santiago-Alarcon et al. Reference Santiago-Alarcon, Palinauskas and Schaefer2012a ). The prevalence, distribution, and diversity of these blood parasites vary among biogeographical regions (Valkiūnas, Reference Valkiūnas2005), altitude (Galen and Witt, Reference Galen and Witt2014; González et al. Reference González, Matta, Ellis, Miller, Ricklefs and Gutiérrez2014), host life history traits (Fecchio et al. Reference Fecchio, Lima, Svensson-Coelho, Marini and Ricklefs2013; Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013; Lutz et al. Reference Lutz, Hochachka, Engel, Bell, Tkach, Bates, Hackett and Weckstein2015; Matthews et al. Reference Matthews, Ellis, Hanson, Roberts, Ricklefs and Collins2016), habitat (Mendes et al. Reference Mendes, Piersma, Lecoq, Spaans and Ricklefs2005; Loiseau et al. Reference Loiseau, Iezhova, Valkiūnas, Chasar, Hutchinson, Buermann, Smith and Sehgal2010, Reference Loiseau, Harrigan, Robert, Bowie, Thomassen, Smith and Sehgal2012), bioclimatic conditions (Oakgrove et al. Reference Oakgrove, Harrigan, Loiseau, Guers, Seppi and Sehgal2014), and can also be affected by urbanization and land use (Evans et al. Reference Evans, Gaston, Sharp, McGowan, Simeoni and Hatchwell2009; Hernández-Lara et al. Reference Hernández-Lara, González-García and Santiago-Alarcon2017). Usually, multiple species of these intracellular parasites coexist locally and exhibit varying levels of host specificity, ranging from infecting a single host to several distantly related hosts (Waldenström et al. Reference Waldenström, Bensch, Kiboi, Hasselquist and Ottoson2002; Fecchio et al. Reference Fecchio, Lima, Svensson-Coelho, Marini and Ricklefs2013; Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013; Drovetski et al. Reference Drovetski, Aghayan, Mata, Lopes, Mode, Harvey and Voelker2014; Ellis et al. Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015). Although some avian malaria lineages might have diverged within a single-host population (Pérez-Tris et al. Reference Pérez-Tris, Hellgren, Križanauskienė, Waldenström, Secondi, Bonneaud, Fjeldså, Hasselquist and Bensch2007), diversification by host switching appears to be common throughout the evolutionary history of this group of parasites (Ricklefs et al. Reference Ricklefs, Fallon and Bermingham2004, Reference Ricklefs, Outlaw, Svensson-Coelho, Medeiros, Ellis and Latta2014; Lauron et al. Reference Lauron, Loiseau, Bowie, Spicer, Smith, Melo and Sehgal2015). These factors listed above may play significant roles in structuring avian malaria parasite assemblages, either within a single-host population or within a more diverse local host community.
Using the mitochondrial cytochrome b (cyt b) gene to delineate parasite lineages, we describe the distribution and diversity of Plasmodium and Haemoproteus in their manakin hosts (Aves: Pipridae) across Central and South America. We focused on this endemic Neotropical bird family as a model system for three reasons: (1) its phylogeny has been relatively well studied (Ohlson et al. Reference Ohlson, Fjeldså and Ericson2013), allowing for detailed investigation of how parasites are related to the evolutionary histories of their hosts; (2) the geographic distributions and diversity of manakins are characterized by high temperature and precipitation (Anciães and Peterson, Reference Anciães and Peterson2009), which may promote high diversity and prevalence of blood parasites through climate influence on vector density and diversity; and (3) manakins are known by their polygynous breeding system, complex courtship displays and strong sexual dimorphism. The clustering of individuals during mating might result in increased exposure of manakins to haemosporidian vectors (Tella, Reference Tella2002; Fecchio et al. Reference Fecchio, Lima, Silveira, Braga and Marini2011) and, under the expectation of significant parasite impact on secondary sexual traits of males (Hamilton and Zuk, Reference Hamilton and Zuk1982), one would predict that manakins are heavily parasitized.
Here, we first asked whether haemosporidian parasite diversity in manakins exhibits turnover among individual host species, and whether this turnover can be explained by the extent of geographic overlap between species and by the degree of phylogenetic relatedness among species. Second, we tested whether turnover in parasite assemblages of the manakin community among sites across Amazonia is related to: (a) geographic distance, (b) turnover in the manakin assemblage, (c) environmental differences and (d) turnover in the bird community of species other than manakins.
MATERIALS AND METHODS
Sample collection
We obtained blood (92% of all samples), or muscle/liver tissue (8%) from 1343 individual birds of 30 species of manakins sampled in four biomes: Amazonia (1027 birds, 24 species), Lowland Tropical Forest (233 birds, four species), Cerrado (57 birds, seven species) and Atlantic Rain Forest (26 birds, one species) (Fig. 1). The birds were netted during different time periods, seasons and/or years. Blood was collected by brachial venipuncture with disposable sterile needles from netted birds. Muscle and liver samples were taken during specimen preparation. All blood samples were stored in 95% ethanol or lysis buffer until DNA extraction. Tissue samples were stored in absolute ethanol or flash frozen in liquid nitrogen. All samples were collected under appropriate permits in Brazil, Ecuador, Panama and Peru.

Fig. 1. Map of sampling localities included in this study. White and black stars represent sites incorporated for analysis 1 (parasite lineage turnover among manakin species). White stars represent localities incorporated in analysis 2 (parasite lineage turnover among localities). Endemic area of Amazonia is shown in parenthesis.
Parasite detection
To screen total DNA samples for presence of Plasmodium and Haemoproteus, we adopted three molecular protocols by using parasite-specific primers targeting a fragment of cyt b. Information for detailed laboratory methods, primers names, and PCR conditions can be found in Fallon et al. (Reference Fallon, Ricklefs, Swanson and Bermingham2003), Hellgren et al. (Reference Hellgren, Waldenström and Bensch2004), Bell et al. (Reference Bell, Weckstein, Fecchio and Tkach2015)]. These screening methods have similar effectiveness to detect haemosporidian parasites (Bell et al. Reference Bell, Weckstein, Fecchio and Tkach2015).
Phylogenetic relationships of parasites
Sequences were edited and the 56 unique haplotypes recovered from manakins were aligned in Geneious v. 7.03 (Biomatters, Ltd., Auckland, New Zealand). For some sequences, the outer reactions amplified using the protocol of Svensson-Coelho et al. (Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013) were then sequenced instead for the nested products. Because different cyt b regions were sequenced by different laboratories, overlap was not complete and thus coverage at individual nucleotide sites across the alignment ranged from 3 to 45 known characters per site (median 36, quartile range 16–45). The shortest sequence was 448 bases long; thus, no sequence included in our alignment was missing more than 46% of the examined sites. The complete alignment lacked data at 35% of the sites. Simulation studies have demonstrated that up to 50% missing data does not pose a problem for assigning placement in phylogenies (Wiens, Reference Wiens2003; Wiens and Morrill, Reference Wiens and Morrill2011). Nonetheless, after estimating an initial maximum-likelihood (ML) phylogeny including all haplotypes, we collapsed shallow clades and repeated the ML analysis, including the one haplotype per clade with more sequence data. If several haplotypes within a clade were of equal length, one was chosen at random for the phylogeny Ohlson et al. (2013). We refer to collapsed clades as putative evolutionary lineages (Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013). Grouping haplotypes in this manner may result in loss of information about geographical or host specificity of parasites: therefore, we examined each lineage more closely in median-joining haplotype networks visualized by the program Network (available at fluxus-engineering.com, Bandelt et al. Reference Bandelt, Forster and Röhl1999). All ML analyses were performed in RA×ML BlackBox (Stamatakis et al. Reference Stamatakis, Hoover and Rougemont2008), applying the default general time reversible (GTR) + gamma model of evolution and running 100 bootstrap replicates.
In a final ML analysis, we incorporated our 39 putative evolutionary lineages and 102 haemosporidian cyt b sequences analysed in Borner et al. (Reference Borner, Pick, Thiede, Kolawole, Kingsley, Schulze, Cottontail, Wellinghausen, Schmidt-Chanasit, Bruchhaus and Burmester2016). Prior to analysis, ends were trimmed to where at least two sequences overlapped to result in a total of 1070 sites being compared. We used the five Leucocytozoon lineages incorporated in the phylogeny of Borner et al. (Reference Borner, Pick, Thiede, Kolawole, Kingsley, Schulze, Cottontail, Wellinghausen, Schmidt-Chanasit, Bruchhaus and Burmester2016) as an outgroup. After analysis, the phylogeny was pruned using the command ‘drop.tip’ in the package ape (Paradis et al. Reference Paradis, Claude and Strimmer2004) in R (R Core Team, 2016) to include only lineages recovered from manakins. To understand the host specificity of the parasite lineages recovered from manakins, we matched parasite lineages to host species association based on data from the local bird communities from which we obtained manakin samples (Fig. 2).

Fig. 2. ML tree of cyt b lineages recovered in manakins. Bootstrap support of ⩾70 is indicated with circles on nodes. The analysis incorporated data from Borner et al. (Reference Borner, Pick, Thiede, Kolawole, Kingsley, Schulze, Cottontail, Wellinghausen, Schmidt-Chanasit, Bruchhaus and Burmester2016) and was subsequently pruned (see text). Lineages from TI P24L to C701 group significantly with other Plasmodium lineages. Lineages SM H8U to TI H17L group significantly within the Haemoproteus clade. Infections from all manakin species are highlighted in dark grey (species codes can be found in Table 1) and other non-manakin species are highlighted in light grey. We also indicate the number of individual non-manakin species (per family) exhibiting infection of a given parasite lineage and the number of species per host family from which the parasite was recovered. In columns on the left, we list number of individuals infected in Pipridae, number of manakin host species utilized, total number of individual host recovered in all sites and hosts combined (including non-manakins), the total number of host species utilized (including non-manakins), and list of the number of endemic areas from which a parasite was recovered (maximum possible = 10). Finally, in rows we list number of individuals infected and number of parasite lineages harboured per host species or family for non-manakins (Richness).
Turnover of malaria parasites
To examine factors potentially influencing the structure of avian malaria parasite assemblages, either among manakin species (Analysis 1) or spatially among local assemblages of manakin species (Analysis 2), we used Mantel and partial Mantel tests (Mantel, Reference Mantel1967; Manly, Reference Manly2007) to obtain correlations and associated significance values. To estimate malaria lineage turnover both between manakin species and among manakin assemblages, we used the Jaccard similarity index (Jaccard, Reference Jaccard1912; Magurran, Reference Magurran2004). To obtain an index that increases with increasing dissimilarity, we subtracted the original Jaccard index from one as follows:
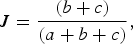 $$J = \displaystyle{{(b + c)} \over {(a + b + c)}},$$
$$J = \displaystyle{{(b + c)} \over {(a + b + c)}},$$
where a is the number of shared species between two assemblages, and b and c are the numbers unique to each assemblage. In all analyses, our operational taxonomic units are manakin species and parasite lineages.
To test for a relationship between parasite turnover among host species and either host phylogenetic relatedness or geographic range overlap, we selected only manakin species in which five or more individuals harboured identified parasite infections (n = 9 species; Fig. 1). We used J to assess turnover between all manakin species pairs. We obtained pairwise host genetic distance from DNA sequences of four genes (ND2, Myoglobin, G3PDH and ODC) from Ohlson et al. (Reference Ohlson, Fjeldså and Ericson2013). We estimated raw pairwise distances and averaged these across the four genes. We obtained geographic range maps of all nine manakin species in our sample from BirdLife International (available from http://www.birdlife.org/datazone/info/spcdownload) and calculated proportional range overlap as ½ (overlap/range size A + overlap/range size B) following Davies and Pedersen (Reference Davies and Pedersen2008).
In the second analysis, in which we sought correlates of turnover in the parasite community among locations, we estimated J not among host species, but among localities. For this analysis, we tested whether malaria turnover among Pipridae assemblages correlates with: (1) turnover of the malaria assemblage of all host species sampled; (2) climatic factors; (3) geographic distance; (4) turnover of the hosts (i.e. of manakin species); or (5) turnover of the overall host assemblage (mostly passerines). At each locality in the study, we sampled non-manakin species following the same protocols for sample collection and identification of infections reported above for manakins. We included only localities in which at least five infections within Pipridae and five infections in the rest of the host assemblage were identified (n = 7 localities in Amazonia with between 91 and 1901 birds sampled, primarily passerines). We refer to the non-manakin bird assemblage as the ‘larger community’ to distinguish it from parasite assemblages within host species (Esch et al. Reference Esch, Bush and Aho1990).
For climate, we obtained 19 bioclimatic variables and elevation from WorldClim (available from http://www.worldclim.org/; Hijmans et al. Reference Hijmans, Cameron, Parra, Jones and Jarvis2005) at a 1 min resolution in DIVA-GIS (available from http://www.diva-gis.org/). We extracted principal components (PCs) from a correlation matrix of these variables, and saved those with an eigenvalue >1. This resulted in three PCs that explained 97% of the variation. Finally, we calculated the Euclidean distances among sites based on these three PCs. We obtained geographic distances in km in DIVA-GIS. Distances between localities are range from 190 km (Comodoro-Brazil and Chupinguaia-Brazil) to 2110 km (Comodoro-Brazil and Tiputini-Ecuador). Finally, we estimated J of the host species assemblages, first for Pipridae only and then for the remaining species. We used the packages vegan (Oksanen et al. Reference Oksanen, Blanchet, Kindt, Legendre, Minchin, O'Hara, Simpson, Solymos, Stevens and Wagner2010), ape (Paradis et al. Reference Paradis, Claude and Strimmer2004), maptools (Bivand et al. Reference Bivand, Lewin-Koh, Pebesma, Archer, Baddeley, Bibiko, Brey, Callahan, Carrillo, Dray, Forrest, Friendly, Giraudoux, Golicher, Gómez-Rubio, Hausmann, Hufthammer, Jagger, Luque, MacQueen, Niccolai, Perpiñán-Lamigueiro, Short, Snow, Stabler, Stokely and Turner2015), PBSmapping (Schnute et al. Reference Schnute, Boers, Haigh, Grandin, Chabot, Johnson, Wessel, Antonio, Lewin-Koh and Bivand2015) and rgdal (Bivand et al. Reference Bivand, Keitt, Rowlingson, Pebesma, Sumner, Hijmans and Rouault2014) in R to calculate all distance matrices.
We estimated both Mantel and partial Mantel correlations (where we test the dependent and one independent variable while controlling for the remaining independent variables) using the package ecodist (Goslee and Urban, Reference Goslee and Urban2007) for R. In all analyses involving our dependent variable (lineage turnover of manakin parasites), we used a one-tailed test. We predicted manakin parasite lineage turnover among host species to correlate negatively with host range overlap and positively with host genetic distance (analysis 1); we predicted manakin parasite lineage turnover among localities to correlate positively with parasite compound community dissimilarity, geographic distance, Pipridae species turnover, climatic dissimilarity and non-Pipridae species turnover.
RESULTS
Haemosporidian prevalence and diversity of lineages in manakins
We analysed 1343 manakins of 30 species sampled across Brazil, Ecuador, Panama and Peru (Fig. 1). Samples were taken from a dataset comprised 5896 individual birds screened for haemosporidian parasites. We detected 146 infections in 22 manakin species with a prevalence of 10·9% (Table 1). Infections varied among host species, ranging from 0 to 50% (Table 1).
Table 1. Blood parasite prevalence (pooled Plasmodium and Haemoproteus) in manakins

Species of birds are organized by prevalence.
a No longer Pipridae.
We recovered 39 haemosporidian cyt b lineages: 23 within the genus Plasmodium and 16 within the genus Haemoproteus (Fig. 2). Fifteen lineages infecting manakins were also found in other bird families (Fig. 2). Twenty-four lineages were found only in manakins, but at this stage we cannot claim that they are all specific to manakins, since we did not sample all the communities where these manakin hosts were caught. However, some rare lineages were found exclusively in manakins within well-sampled bird communities. For example, lineage C701L was found in only a single fiery-capped manakin (Machaeropterus pyrocephalus) individual in our Peru site, where we also screened 717 birds belonging to 115 species. The lineage P16L found exclusively in six manakin species across four areas of endemism in Amazonia and Cerrado represents the most widespread and specialized lineage of manakin parasite.
Turnover of lineages
Among Pipridae host species, malaria parasite lineage turnover ranged from 0·0 to 0·43 (mean 0·12 ± 0·11 s.d.; Table 2). This variation did not correlate with either manakin species range overlap or genetic divergence between hosts (Fig. 3). Among Amazonian localities, malaria lineage turnover ranged from 0·0 to 0·40 (mean 0·16 ± 0·12 s.d.; Table 3). This variation did not correlate with manakin species turnover, geographic distance, climate difference, species turnover in the larger bird community or lineage turnover of parasites recovered from non-manakin species (Fig. 4). Some independent variables were significantly correlated with each other, although the correlation coefficient never exceeded 0·6 (Table 4).

Fig. 3. Lineage turnover in avian malaria assemblages among nine manakin host species in relation to host genetic distance (A) and geographic range overlap (B). Mantel correlation coefficients (r) and one-tailed significance thereof (P) are reported. Partial Mantel test, where we control for the second independent variable, are presented in parentheses.

Fig. 4. Lineage turnover in avian malaria assemblages among seven Amazonian localities in relation to: turnover of malaria lineages in non-manakin species (A); turnover among the non-manakin bird assemblages (B); climatic differences (C); turnover among manakin assemblages (D); and geographic distance (E). Mantel correlations and significance thereof are reported. Partial Mantel correlations (r), in which the remaining four independent variables are controlled for, are shown in parentheses.
Table 2. Manakin malaria parasite lineage turnover estimated by the Jaccard index [equation (1)] among nine host species. Species names can be found in Table 1

Table 3. Manakin malaria parasite lineage turnover estimated by the Jaccard index [equation (1)] among seven Amazonian localities (Fig. 1). Comparisons within two endemic areas are highlighted in different shades of grey

Table 4. Correlations between independent variables used in calculating among-locality turnover of avian malaria parasites of Pipridae. Independent variables considered are: (1) parasite assemblage turnover within the rest of the bird assemblage (CdC); (2) climatic differences (Clim); (3) geographic distance (Geo); (4) Pipridae assemblage turnover (Pip); and (5) turnover of the larger avian assemblage (Bird). Two-tailed P values are reported

DISCUSSION
We investigated broad ecological and evolutionary factors that may shape the diversity and distribution of haemosporidian parasites in manakins across the Neotropics. In our analyses, we found low to moderate levels of lineage turnover across manakin species and localities. However, none of the seven factors that we tested explained variation in malaria parasite lineage turnover in this host family.
A diminishing proportion of shared parasites among host communities with increasing geographic distance has been found in several host–parasites systems such as helminthes of freshwater fish and mammals (Poulin, Reference Poulin2003), mites and fleas from small mammals (Krasnov et al. Reference Krasnov, Shenbrot, Mouillot, Khokhlova and Poulin2005; Vinarski et al. Reference Vinarski, Korallo, Krasnov, Shenbrot and Poulin2007), trematodes from marine molluscs (Thieltges et al. Reference Thieltges, Ferguson, Jones, Krakau, Montaudouin, Noble, Reise and Poulin2009) and malaria parasites from birds across island archipelagoes (Ishtiaq et al. Reference Ishtiaq, Clegg, Phillimore, Black, Owens and Sheldon2010; Svensson-Coelho and Ricklefs, Reference Svensson-Coelho and Ricklefs2011). Our results do not agree with these previous findings. One plausible explanation for this is that haemosporidian dispersal in continental ecosystems is not as constrained as it is in insular ecosystems. For example, the turnover of haemosporidian parasites across bird populations in eastern North America (Ellis et al. Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015) and high degree of similarity in diptera vectors species across landscapes with different land use in Mexico (Abella-Medrano et al. Reference Abella-Medrano, Ibáñez-Bernal, MacGregor-Fors and Santiago-Alarcon2015; Hernández-Lara et al. Reference Hernández-Lara, González-García and Santiago-Alarcon2017) demonstrate the capability of these parasites to disperse within regions and to switch between avian hosts. Here, we show similarly low turnover of haemosporidia across a large geographic region in the Neotropics.
Evidence for that malaria parasite community structure relates to that of birds is mixed. Whereas Svensson-Coelho and Ricklefs, (Reference Svensson-Coelho and Ricklefs2011) found no evidence for the predicted positive association between bird and malaria assemblage dissimilarity across islands in the Caribbean, Ellis et al. (Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015) did in their continental system in eastern North America. Here, we found no evidence for such an association. This is true both when considering only the manakin assemblage and when considering the larger bird assemblage. An explanation for the lack of such association is that the distribution of haemosporidia is constrained by vector distribution. Although we have no information about vectors in our study region, Ishtiaq et al. (Reference Ishtiaq, Guillaumot, Clegg, Phillimore, Black, Owens, Mundy and Sheldon2008) argued that the movement of avian malaria lineages between southwest Pacific Islands might be restricted by the lack of distributional overlaps of competent vector species.
A positive correlation between parasite sharing and host geographical overlap is expected because opportunities for host shifting should increase where a non-infected potential host species occurs in sympatry with an actual host species (Fenton and Pedersen, Reference Fenton and Pedersen2005; Davies and Pedersen, Reference Davies and Pedersen2008; Ricklefs et al. Reference Ricklefs, Outlaw, Svensson-Coelho, Medeiros, Ellis and Latta2014). Indeed, this has been found for several types of parasites and pathogens in wild primates and humans (Davies and Pedersen, Reference Davies and Pedersen2008). However, here we show that turnover among local assemblages of haemosporidian parasites of manakins is largely insensitive to host range overlap, 19 bioclimatic variables, and the dissimilarity of alternative hosts (i.e. non-manakin).
Environmental factors have been shown to affect the prevalence, distribution and alpha diversity (Gonzalez-Quevedo et al. Reference Gonzalez-Quevedo, Davies and Richardson2014; Oakgrove et al. Reference Oakgrove, Harrigan, Loiseau, Guers, Seppi and Sehgal2014; Olsson-Pons et al. Reference Olsson-Pons, Clark, Ishtiaq and Clegg2015), but not beta diversity of avian malaria parasites (Svensson-Coelho and Ricklefs, Reference Svensson-Coelho and Ricklefs2011; Ellis et al. Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015). Malaria parasites are never subjected to the external environment directly, but they do pass through an ectothermic vector as part of their life cycle. At that time, they may be indirectly exposed to the external environment. For example, lower temperature not only constraints the sporogonic development of these parasites, but also reduces the activity of its mosquito vectors (Valkiūnas, Reference Valkiūnas2005), which consequently could influence transmission and prevalence. Our study, however, is set in a tropical ecosystem, where temperature and moisture are unlikely to limit parasite transmission. Hence, the slight variation in climate across sites may have no consequence for Amazonian haemosporidians.
The lack of an association between malaria lineage turnover and all independent dissimilarity variables examined here supports the idea that local coevolution between avian hosts and avian malaria parasites, as well as local vector ecology, are more important in structuring malaria assemblages than distance–decay relationships (Svensson and Ricklefs, Reference Svensson-Coelho and Ricklefs2011; Scordato and Kardish, Reference Scordato and Kardish2014). Additionally, transmission of multi-host pathogens, such as avian malaria, seems to be driven mostly by heterogeneity in host–parasite compatibility (Medeiros et al. Reference Medeiros, Hamer and Ricklefs2013), thereby making external factors (other potential host species, climate, geography, etc.) less influential. External factors may nonetheless influence malaria assemblages to some degree, which would explain the significant associations recovered in some studies (e.g. Ishtiaq et al. Reference Ishtiaq, Clegg, Phillimore, Black, Owens and Sheldon2010; Ellis et al. Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015)
Host switching has been shown to be a central event in the diversification of haemosporidians in New World passerine (Ricklefs et al. Reference Ricklefs, Outlaw, Svensson-Coelho, Medeiros, Ellis and Latta2014) and non-passerine birds (Santiago-Alarcon et al. Reference Santiago-Alarcon, Rodríguez-Ferraro, Parker and Ricklefs2014) and is reflected in patterns recovered in this study in that 15 of 39 (38%) lineages recovered from manakins are also found in non-manakin hosts. The diversity of bird families within Amazonia and movement of haemosporidian lineages among these families must have played a role in forming the high diversity within this group of parasites in the Neotropics. The definitive hosts (i.e. vectors from the families Ceratopogonidae and Culicidae, respectively) of both Haemoproteus and Plasmodium have been poorly studied with regard to distribution, density and feeding preferences, and, in most cases, competent parasite vectors have not been identified (Santiago-Alarcon et al. Reference Santiago-Alarcon, Palinauskas and Schaefer2012a ). Recent studies on feeding preferences of blood-sucking diptera have revealed low preference for specific bird species, suggesting that vectors do not present barriers between bird and parasite encounters (e.g. Santiago-Alarcon et al. Reference Santiago-Alarcon, Havelka, Schaefer and Segelbacher2012b ; Medeiros et al. Reference Medeiros, Ricklefs, Brawn and Hamer2015; Ferreira et al. Reference Ferreira, Rodrigues, Sato, Borges and Braga2016). Further investigation into parasite vector distribution, host specificity and transmission will undoubtedly help to determine whether haemosporidian lineages track the diversity of their definitive hosts or of their intermediate hosts across Neotropical region.
ACKNOWLEDGEMENTS
We thank all ornithologists and field assistants who helped collect the samples. We also thank the Bird Collection of Instituto Nacional de Pesquisas da Amazônia, Museu Paraense Emílio Goeldi, Renato Caparroz and Milene Gaiotti who loaned part of the samples used in this study.
FINANCIAL SUPPORT
This work was funded by the Fundação de Amparo à Pesquisa do Estado do Amazonas – FAPEAM and Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (Process number 61·0012/2008·7-DCR/CNPq) to A.F., National Science Foundation DEB-1503804 to J.D.W., DEB-1120734 to V.T. and DEB-1146491 to C.C.W. During the project, MS-C received support from the São Paulo Research Foundation (process numbers 2012/08576-6 and 2015/17523-1) and A.F. received a Postdoctoral Fellowship from CNPq and FAPEAM (Process numbers 350140/20120 and 201275/2014-7). A.F. is currently maintained by a PNPD scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).








