


Drug-resistant epilepsy is defined as the continuation of debilitating seizures, despite adequate therapy with two or more antiepileptic medications alone or in combination.Reference Berg, Berkovic and Brodie 1 The most severe forms of drug-resistant epilepsy usually begin in infancy and are associated with global developmental delay, cognitive dysfunction, and ongoing epileptiform activity, which cause further cognitive slowing and decline, defined as epileptic encephalopathy.Reference Berg, Berkovic and Brodie 1 Half of these children are candidates for ketogenic diet therapy for seizure management.
Ketogenic diets have been used since the 1920s for the treatment of intractable epilepsy.Reference Kossoff, Zupec-Kania and Amark 2 There are only two inherited metabolic disorders for which the effect of the ketogenic diet on seizure control is well known: (1) glucose transporter 1 (GLUT1) deficiency, which is caused by likely pathogenic variants in SLC2A1;Reference Wang, Pascual and De Vivo 3 and (2) pyruvate dehydrogenase complex deficiency, which results in decreased acetyl-CoA and ATP production.Reference Wexler, Hemalatha and McConnell 4 In both disorders, a ketogenic diet provides an alternative fuel source to the brain and bypasses glucose metabolism.
Discontinuation of the ketogenic diet will certainly result in seizure reoccurrence and deterioration of cognitive functions in patients with GLUT1 deficiency. Confirmed diagnosis of GLUT1 deficiency will likely prevent the negative effects of early discontinuation of the ketogenic diet. To identify patients with GLUT1 deficiency and be able to decide on the duration of the ketogenic diet therapy, we performed a prospective cohort study to investigate GLUT1 deficiency by targeted SLC2A1 genetic testing using direct sequencing and multiplex ligation probe amplification (MLPA) of SLC2A1 in patients with drug-resistant epilepsy treated with the ketogenic diet. To investigate the prevalence of GLUT1 deficiency and other genetic diagnoses in epilepsy patients on the ketogenic diet, we also retrospectively reviewed patients for their diagnosis in the same clinic within the same period as our prospective cohort study.
The prevalence of GLUT1 deficiency prospective cohort study was approved by the institutional research ethics board (no. REB#1000033443). All patients with drug-resistant epilepsy on the ketogenic diet with no genetic diagnosis were approached for this prospective cohort study between July of 2012 and December of 2014 at a single ketogenic diet clinic. DNA samples of enrolled patients with signed consents underwent direct sequencing (all exons and intron boundaries), and MLPA of the SLC2A1 for deletions and duplications at the Metabolic Laboratory in Amsterdam (the Netherlands) according to their standard diagnostic laboratory methods.
The prevalence of genetic diagnoses in patients with drug-resistant epilepsy on the ketogenic diet as a retrospective cohort study for the patients seen between July of 2012 and December of 2014 at the same ketogenic diet clinic was also approved by the institutional research ethics board (no. REB#1000049758). We reviewed electronic patient charts for clinical features, neuroimaging studies, biochemical investigations, and molecular genetic investigations, including targeted next-generation sequencing panels of epileptic encephalopathy. All information was entered into an Excel database.
Thirty-nine patients underwent prospective targeted direct sequencing and MLPA of SLC2A1. None of these patients had a pathogenic variant in SLC2A1.
There were 99 patients in the retrospective cohort study, including the 39 patients from the prevalence of GLUT1 deficiency prospective cohort study. There were 37 females and 62 males. The current average age was 8 years and 10 month ±5 years and 5 month standard deviation (range 1 year and 6 months-18 years). The average age of seizure onset was 20.14 ±30.83 months (range=1-168 months).
The clinical features, seizure types, number of antiepileptic medications, and neuroimaging features are summarized in Table 1. The most common seizure type (74%) was generalized tonic–clonic (Table 1). Fifty percent of patients had more than one type of seizure, 7% had a movement disorder, and 13% had a behavioral disorder.
Table 1 Clinical features and neuroimaging results of 99 patients on the ketogenic diet
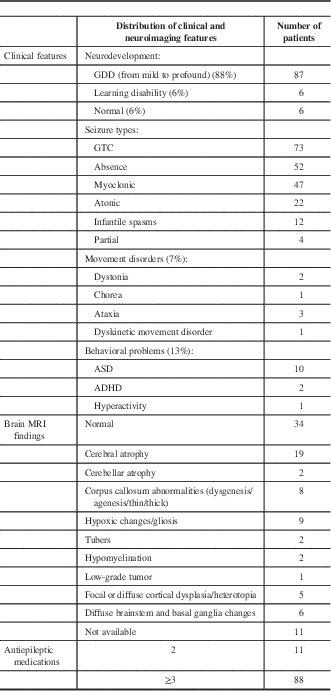
ADHD=attention deficit hyperactivity disorder; ASD=autism spectrum disorder; GDD=global developmental delay; GTC=generalized tonic–clonic; MRI=magnetic resonance imaging.
The majority of patients (89%) had generalized spike or spike-and-wave electrical activity in their electroencephalography (EEG). Eighty-nine percent of patients underwent brain magnetic resonance imaging (MRI). Brain MRI was abnormal in 66% of these patients (Table 1).
Eighty-nine percent of patients were tried on three or more antiepileptic medications prior to the ketogenic diet therapy. A small number of patients received more than 10 antiepileptic medications.
A total of 14 of the 99 patients had no genetic testing and also declined to participate in the prospective GLUT1 study. We excluded these patients from the prevalence calculations. Eleven patients were diagnosed by targeted genetic testing or microarray including GLUT1 deficiency (n=2, published previouslyReference Mercimek-Mahmutoglu, Patel and Cordeiro 5 , Reference Almuqbil, Go, Nagy, Pai, Mamak and Mercimek-Mahmutoglu 6 ), pathogenic copy number variants (n=3), congenital disorders of glycosylation (n=1), neuronal ceroid lipofuscinosis type II (n=1), mitochondrial disorder by mutations in the NDUFS7 (n=1), tuberous sclerosis (n=2), and lissencephaly by mutation in the LIS1 (n=1) (Table 2). Seven patients were diagnosed by targeted next-generation sequencing panels of epileptic encephalopathy genes, including SCN1A- (n=4), SCN8A- (n=1), and STXBP1- (n=2) associated epileptic encephalopathies (Table 2). The prevalence of a genetic diagnosis was 21% (18/85) in our retrospective cohort study.
Table 2 All patients with a genetic diagnosis
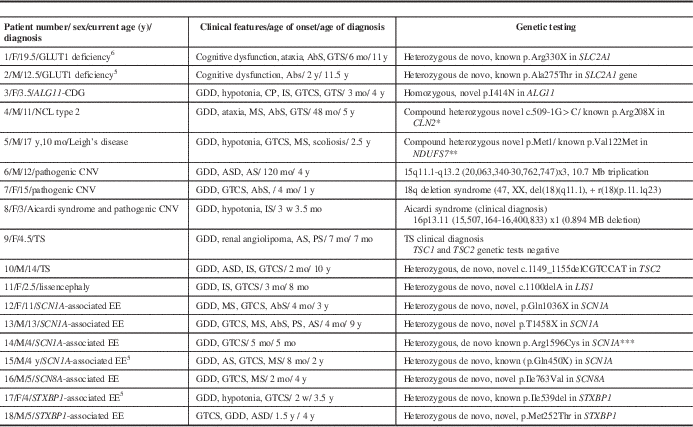
* Sleat DE, Donnelly RJ, Lackland H, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277(5333):1802-5.
** Triepels RH, Van den Heuvel LP, Loeffen JL, et al. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45(6):787-90.
*** Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(Pt 3):843-52.
AbS=absence seizures; ADHD=attention deficit hyperactivity disorder; AS=atonic seizures; ASD=autism spectrum disorder; CDG=congenital disorders of glycosylation; CNV=copy number variants; CP=cerebral palsy; EE=epileptic encephalopathy; F=female; GCS=generalized clonic seizures; GDD=global developmental delay; GTCS=generalized tonic–clonic seizures; GTS=generalized tonic seizures; GLUT1=glucose transporter 1; IS=infantile spasms; M=male; mo=month(s); MS=myoclonic seizures; PS=partial seizures; TS=tuberous sclerosis; w=week(s); y=year(s).
In patients with drug-resistant epilepsy on the ketogenic diet, the prevalence of GLUT1 deficiency was 2.4%, and the prevalence of a genetic diagnosis was 21% in the single ketogenic diet clinic. The SCN1A-associated epileptic encephalopathy was the most common genetic diagnosis in our study. We did not identify any new patients with GLUT1 deficiency, which could be due to the small number of patients enrolled in the prospective cohort study.
The prevalence of GLUT1 deficiency in epilepsy patients on the ketogenic diet was between 0 and 8.5% in three studies.Reference Becker, Schubert and Weckhuysen 7 - Reference Schoeler, Cross and Drury 9 Those studies included a total of 307 patients (274 using direct sequencing only and 33 direct sequencing and MLPA of SLC2A1).Reference Becker, Schubert and Weckhuysen 7 - Reference Schoeler, Cross and Drury 9 Only one patient was diagnosed with GLUT1 deficiency.Reference Schoeler, Cross and Drury 9 Including our prospective study results, so far 346 patients have undergone targeted SLC2A1 genetic testing, and the prevalence of GLUT1 deficiency was 0.29% in patients with drug-resistant epilepsy on the ketogenic diet. There is only one retrospective study that looked at the frequency of GLUT1 deficiency in patients on the ketogenic diet.Reference Ramm-Pettersen, Nakken, Haavardsholm and Selmer 8 That study reported occurrence of GLUT1 deficiency in 11 of 130 patients (8.5%). Including our retrospective study results, so far 13 of 215 patients with drug-resistant epilepsy on the ketogenic diet (6%) were diagnosed with GLUT1 deficiency.Reference Ramm-Pettersen, Nakken, Haavardsholm and Selmer 8 Comparison of the prospective (0.29%) and retrospective (6%) prevalence data of GLUT1 deficiency raises the question of whether GLUT1 deficiency is a rare disease. To investigate the prevalence of GLUT1 deficiency in patients with drug-resistant epilepsy on the ketogenic diet, we should be enrolling larger numbers of patients. However, we think that the small number of studies (including our current study) gives valuable and sufficient information for the prevalence of GLUT1 deficiency in patients with drug-resistant epilepsy on the ketogenic diet.
There are only two studies reporting a 14% prevalence of genetic diagnoses in patients on the ketogenic diet.Reference Becker, Schubert and Weckhuysen 7 , Reference Schoeler, Cross and Drury 9 In one of those studies, SCN1A-associated epileptic encephalopathy was the most common genetic diagnosis in 5.6% of patients.Reference Schoeler, Cross and Drury 9 Our study reports a 21% prevalence of a genetic diagnosis in the ketogenic diet clinic. SCN1A-associated epileptic encephalopathy was the most common genetic diagnosis (22%) in our retrospective cohort study. Targeted next-generation sequencing panels of epileptic encephalopathy genes confirmed a genetic diagnosis in 39% of the patients in our study, which is the likely cause of the slightly higher prevalence of genetic diagnoses in our study. Despite the fact that SLC2A1 is included in the targeted next-generation sequencing panels of epileptic encephalopathy genes, we did not identify any patients with GLUT1 deficiency in 93 patients who underwent this genetic testing in our recent study.Reference Mercimek-Mahmutoglu, Patel and Cordeiro 5 We think that GLUT1 deficiency might likely be a rare genetic disease.
Good responses to the ketogenic diet raises a suspicion of GLUT1 deficiency.Reference Wang, Pascual and De Vivo 3 , Reference Pong, Geary, Engelstad, Natarajan, Yang and De Vivo 10 Seizure freedom was reported in 67% of the patients (41/61) with GLUT1 deficiency in one study.Reference Pong, Geary, Engelstad, Natarajan, Yang and De Vivo 10 In another study, seizure freedom was found in 90% of patients (10/11) with GLUT1 deficiency, whereas only 9% of the responders with no diagnosis were seizure-free.Reference Becker, Schubert and Weckhuysen 7 The authors concluded that seizure freedom is an important clue to the diagnosis of GLUT1 deficiency. However, another study reported <8% seizure freedom in patients on the ketogenic diet.Reference Schoeler, Cross and Drury 9 The authors did not think that seizure freedom was solely related to the GLUT1 deficiency.Reference Schoeler, Cross and Drury 9 In our study, both patients with GLUT1 deficiency, treated after 10 years of age, did not become seizure-free. One patient with no response had compliance problems,Reference Almuqbil, Go, Nagy, Pai, Mamak and Mercimek-Mahmutoglu 6 and another patient with good compliance had occasional seizures. It is not clear if a response to the ketogenic diet is solely dependent on compliance or that there are other factors that influence response to the ketogenic diet in patients with GLUT1 deficiency.
In summary, we report a wide spectrum of genetic disorders in patients with drug-resistant epilepsy on the ketogenic diet. The prevalence of genetic diagnoses was 21%, and the prevalence of GLUT1 deficiency was 2.4% in patients with drug-resistant epilepsy on the ketogenic diet. We believe that targeted next-generation sequencing panels of epileptic encephalopathy genes and clinical whole exome sequencing will likely increase the number of patients with GLUT1 deficiency to be treated with the ketogenic diet.
Acknowledgments
We would like to thank the referring physicians. We would also like to thank Helen Lowe for informing parents about this study. We would like to thank Yeoumei Christiana Liu and Helen Lowe for application of the ketogenic diet. We would like to thank the patients and parents for participating in this research study. As principal author of this study, Dr. Mercimek-Andrews would like to thank Dr. Berge Minassian for his early support and encouragement in the development of her research program and for his ongoing guidance and mentorship. This study was partially funded by the Rare Disease Foundation Microgrant Program (November 2011). We would also like to thank the Rare Disease Foundation for their great support. Dr. Mercimek-Andrews was funded through the Department of Pediatrics at the University of Toronto (new investigator start-up funding), and we would like to thank them for this support.
Disclosures
Stacy Hewson, Ledia Brunga, Matilde Fernandez Ojeda, Elizabeth Imhof, Jaina Patel, Maria Zak, Elizabeth J. Donner, Jeff Kobayashi, Gajja S. Salomons, and Saadet Mercimek-Andrews hereby declare that they have no conflicts of interest to disclose.
Statement of Authorship
SH was responsible for patient enrollment, data analysis, arrangement of research samples, and drafting and finalizing of the manuscript. LB, JP, and EI were responsible for data entry and data analysis. MFO was responsible for direct sequencing and MLPA of SLC2A1 for the research study. MZ, EJD, and JK were responsible for application of the ketogenic diet, patient enrollment, and revision and final approval of the manuscript. GSS was responsible for analysis of the results of direct sequencing and MLPA of SLC2A1 for the research study, and revision and final approval of the manuscript. SMA was responsible for design and conceptualization of the study, analysis and interpretation of the data, and drafting and finalizing the manuscript for intellectual content.


