


Background
Primary mitochondrial diseases are a heterogeneous group of rare genetic disorders that can impair the respiratory transport chain, structural components of proteins in mitochondrial membranes, and mitochondrial DNA maintenance.Reference Vafai and Mootha1 As several different protein complexes are responsible for the formation, regulation, and function of the respiratory chain, there are a vast number of nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) mutations that can contribute to clinical mitochondrial disorders.Reference Chinnery2–Reference Alston, Rocha, Lax, Turnbull and Taylor4 There are a number of well-known clinical mitochondrial syndromes, such as mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) and myoclonic epilepsy and ragged-red fiber disease (MERRF), which both arise from heteroplasmic point mutations in mtDNA; or Leigh syndrome, which can occur due to either mitochondrial or nuclear gene mutations.Reference Zeviani and Di Donato5 Mitochondrial disorders typically affect multiple systems, particularly those whose functions are highly dependent on aerobic metabolism, such as the nervous and muscular systems.Reference Pfeffer, Majamaa, Turnbull, Thorburn and Chinnery6 Possible clinical features include neurological manifestations such as ataxia, optic neuropathy, and seizures, and multi-systemic features such as cardiomyopathy, hearing loss, myopathy, ophthalmoplegia, gastrointestinal dysmotility and function, and metabolic acidosis.Reference Zeviani and Di Donato5 Although evidence regarding the epidemiology of mitochondrial disorders is sparse, studies in Australia and England estimated a prevalence of clinically diagnosed disease of at least 1 in 8000 to 1 in 16,000,Reference Skladal, Halliday and Thorburn7–Reference Gorman, Schaefer and Ng9 with a higher prevalence of the presence of pathogenic mtDNA or nDNA mutations.Reference Gorman, Schaefer and Ng9
Due to the heterogeneity of the clinical features of different mitochondrial disorders, the specific interventions used to diagnose and treat patients vary widely and different medical specialties, most notably neurologists or geneticists, provide diagnostic care and ongoing disease management support.Reference Parikh, Goldstein and Koenig10 Few disease-specific interventions are available; examples of treatments for mitochondrial diseases include exercise and diet regimens, and vitamin “cocktails,” with evidence of variation in treatment among providers.Reference Parikh, Goldstein and Koenig11 The Mitochondrial Medicine Society (MMS) recently developed and published guidelines for the diagnosis and management of mitochondrial diseasesReference Parikh, Goldstein and Koenig12 and consensus standards for the ongoing care of diagnosed patients.Reference Parikh, Goldstein and Karaa13 The guidelines were informed by a systematic review of existing literature, and both the guidelines and care standards were developed based on consensus methods involving experts in mitochondrial medicine. While these recommendations are likely to result in greater standardization of care for mitochondrial diseases, the authors acknowledge that much of this guidance was not evidence-based, in part due to the low prevalence of the individual mitochondrial disease syndromes, which creates challenges in generating robust evidence to inform best practices.
In order to improve diagnosis and treatment for patients with mitochondrial diseases, there is an initial need to better understand the organization of care. Understanding what care is currently being provided and where there is heterogeneity in practice will help to support benchmarking against published guidelines and standards and to establish priorities for formal evaluative research designed to determine which interventions are associated with best outcomes for patients. Accordingly, we conducted a cross-sectional survey of Canadian physicians involved in the diagnosis and/or provision of ongoing care to patients with mitochondrial diseases. The three main objectives of this study were as follows:
(i) to describe care for patients with mitochondrial diseases in Canada, with respect to the providers involved and services provided;
(ii) to investigate variation in care, with respect to diagnostic tests, patient monitoring, and management/treatment; and
(iii) to describe Canadian clinicians’ views on research priorities for patients with mitochondrial diseases.
Methods
The study protocol was approved by the Ottawa Health Sciences Network Research Ethics Board (Protocol # 20160721-01H) and by the Children’s Hospital of Eastern Ontario Research Ethics Board (Protocol # 16/133X).
Sample Selection and Survey Implementation
Our study population encompassed Canadian physicians with interest and expertise in diagnosing and/or treating mitochondrial disease. Because neither a single specialty nor professional association comprehensively represents this group, we used snowball sampling as the main method of participant identification and recruitment.Reference Sadler, Lee, Lim and Fullerton14 To develop the initial sampling frame, we formed an advisory group of Canadian experts in mitochondrial medicine who identified names of potential respondents based on their professional networks. We also sampled from relevant professional organizations and research networks with publicly available membership lists, such as the American College of Medical Genetics and Genomics and the Canadian Inherited Metabolic Diseases Research Network. As part of survey completion, participants in the initial sample were asked to suggest the names of potentially eligible and interested colleagues, who then formed the snowball sample. They in turn had the option to recommend other mitochondrial disease care providers for recruitment in their responses.
Potential participants were invited to participate in the survey by mail and could complete the survey by mail or internet.Reference VanGeest, Johnson and Welch15, Reference Hocking, Lim, Read and Hellard16 To invite their participation, sampled physicians were contacted up to five times in an approach modified from Dillman.Reference Dillman, Smyth and Christian17 These contacts were spread out over a period of several weeks and included: a pre-notification letter about the study; a survey package (cover letter, questionnaire booklet, $5 coffee shop gift card, and postage-paid return envelope); a reminder letter for non-respondents; and up to two further survey packages (without the gift card) to remaining non-respondents. The cover letter included with the survey package also provided a link to the online version of the survey with an access code unique to the respondent. The online version of the questionnaire was implemented using a secure Research Electronic Data Capture (REDCap)Reference Harris, Taylor, Thielke, Payne, Gonzalez and Conde18 database housed at the Children’s Hospital of Eastern Ontario. The survey, including multiple rounds of snowball sampling, was conducted between December 2016 and July 2017.
Questionnaire
The questionnaire was developed by the study investigators with some questions about demographics and practice characteristics adapted from the National Physician Survey.19 The survey addressed the following topics: respondents’ personal experience in care for patients with mitochondrial disorders; their views regarding diagnostic, treatment, and care coordination/monitoring practices; their research interests and priorities; and their personal and practice characteristics.
Data Analysis
Paper survey data were entered into REDCap using double data entry by experienced data clerks; discrepancies were identified and resolved by the first author. We conducted descriptive analysis, reporting proportions with 95% confidence intervals. We were interested in identifying tests and treatments characterized by high variability in their perceived usefulness among physicians; following a previous publication,Reference Potter, Little and Chakraborty20 we defined an intervention as having high variability when 20% or more of respondents indicated that they would find the intervention helpful in “all or most” cases, and 20% or more of respondents indicated that they would find the intervention helpful “in rare cases” or “never.” Responses to selected open-ended questions were coded by one researcher (KP) and verified by a second researcher (BKP). In some cases, to avoid reporting identifying information about participants, responses were grouped into categories representing the diverse regions of Canada including the west coast, the Prairie Provinces, central Canada, and the Atlantic region.
Results
Response Rate and Sample Characteristics
From our initial sample of 90 physicians, we obtained the names of an additional 22 physicians through snowball sampling. We received 58 responses (response rate: 58/112 = 52%). Among the 58 responses, 47 were returned by mail and 11 were completed online. Forty-seven respondents (81%) provided useable data and reported an interest in the diagnosis or management of mitochondrial disorders and thus completed the entire survey. The remainder of the results are based on the responses of those 47 participants.
Nearly all respondents reported having an active clinical practice, with 89% spending any of their practice time engaged in the care of patients with mitochondrial disorders (Table 1). Of participants spending any time engaged in caring for patients with mitochondrial disorders, 83% reported spending 20% or less of their clinical practice time, and 100% reported spending 50% or less of their clinical practice time caring for patients with mitochondrial disorders. Participants’ ages ranged from 35 to over 65 years and they reported practicing mainly in universities or academic health sciences centers, in seven different provinces in Canada. Most (84%) reported practicing in urban/suburban settings (Table 1). The majority of respondents identified themselves as specialists in medical genetics (72%) with general pediatrics as the next most common specialty (40%).
Table 1: Sample characteristics (N = 47)*
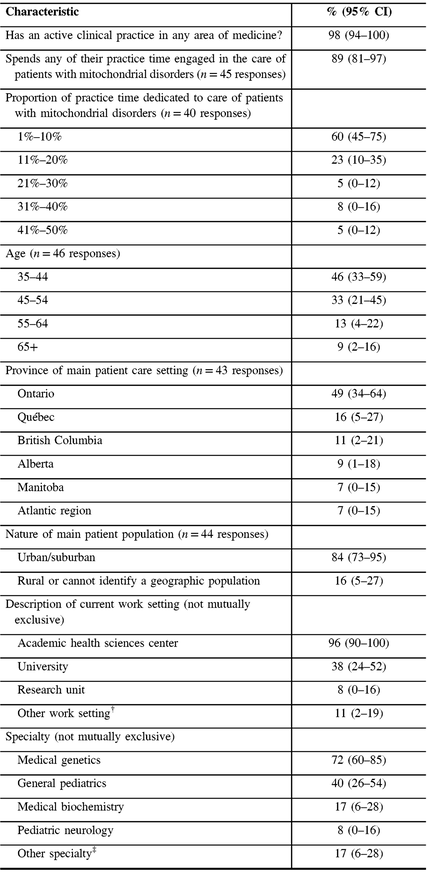
CI = confidence interval.
* Not all proportions listed are out of 47, due to the variable number of responses for each item.
† Other work setting includes community health center, private office/clinic, non-academic health sciences center teaching hospital, community hospital.
‡ Other specialty includes Adult Neurology, Diagnostic Radiology, Pediatric Radiology, Neuroradiology, Adult Nephrology, Pediatric Nephrology, Pain Medicine, Pediatric Critical Care Medicine, Internal Medicine, Adult Endocrinology and Metabolism, Pediatric Endocrinology and Metabolism, and Physical Medicine and Rehabilitation.
Clinical Experience with Mitochondrial Disorders
The vast majority of participants (37/42 responding to the question) reported providing care for patients with any type of mitochondrial disorder in the past 12 months. Non-classical but clearly mitochondrial (based on clinical and/or laboratory features) disorders were reported as the most commonly treated disorders within the past year (83%) (Table 2). Other disorders treated by a majority of respondents in the past year were MELAS (69%), Leigh-like disease (60%), congenital lactic acidosis (56%), and chronic progressive external ophthalmoplegia syndrome (CPEO) (52%). All disorders we asked about had been treated by at least 16% of the respondents in the past year or past 5 years (Table 2).
Table 2: Respondents’ experience in treating mitochondrial disorders (N = 47)*
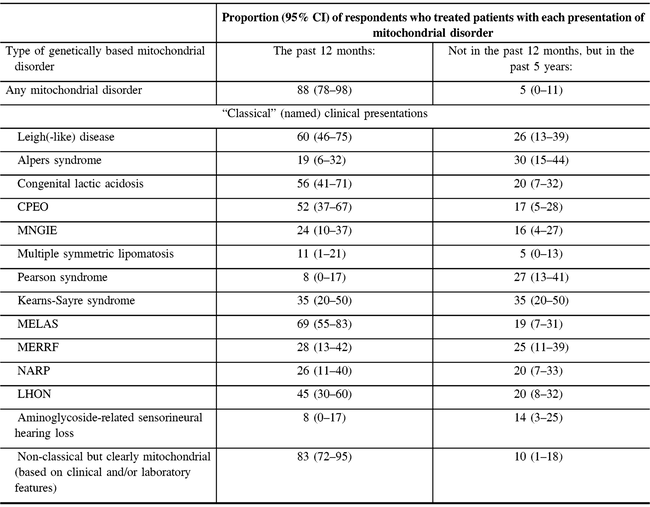
MNGIE = myoneurogastrointestinal disorder and encephalopathy; NARP = neuropathy, ataxia, and retinitis pigmentosa; LHON = Leber’s hereditary optic neuropathy.
* Not all proportions listed are out of 47, due to the variable number of responses for each item. Valid responses were received from at least 36 and up to 43 responses for each type of disorder.
Diagnostic Practices
Nearly all participants who completed the survey reported having provided a diagnostic workup for a patient suspected to have a mitochondrial disorder within the past 5 years, and about half (53%) reported following a specific diagnostic algorithm.
Participants were asked to evaluate various diagnostic investigations according to how diagnostically helpful they were considered to be, i.e., in all or most cases, in some cases, in rare cases, or never (respondents could also select “would be helpful but not available”; these responses were reported separately and removed from the denominator when reporting on views about helpfulness). The only non-genetic diagnostic investigation that was endorsed as “helpful in all or most cases” by a majority of participants was brain magnetic resonance imaging (MRI)/MR spectroscopy (61%); the next most endorsed non-genetic assessment was lactate (44%) (Figure 1). Based on our a priori definition of “high variability” in physician endorsement (see Methods), blood gases, acylcarnitine profile, skin biopsy for fibroblast culture, and one muscle biopsy technique (Bergstrom needle) were assessments with high variability in physician ratings of helpfulness (Figure 1). No non-genetic diagnostic assessments were reported as “potentially helpful but not available” by a majority of respondents (not shown in figure). The two assessments most commonly reported as unavailable were both muscle biopsy assessments: suction needle (Bergstrom) (34%) and needle biopsy (47%).
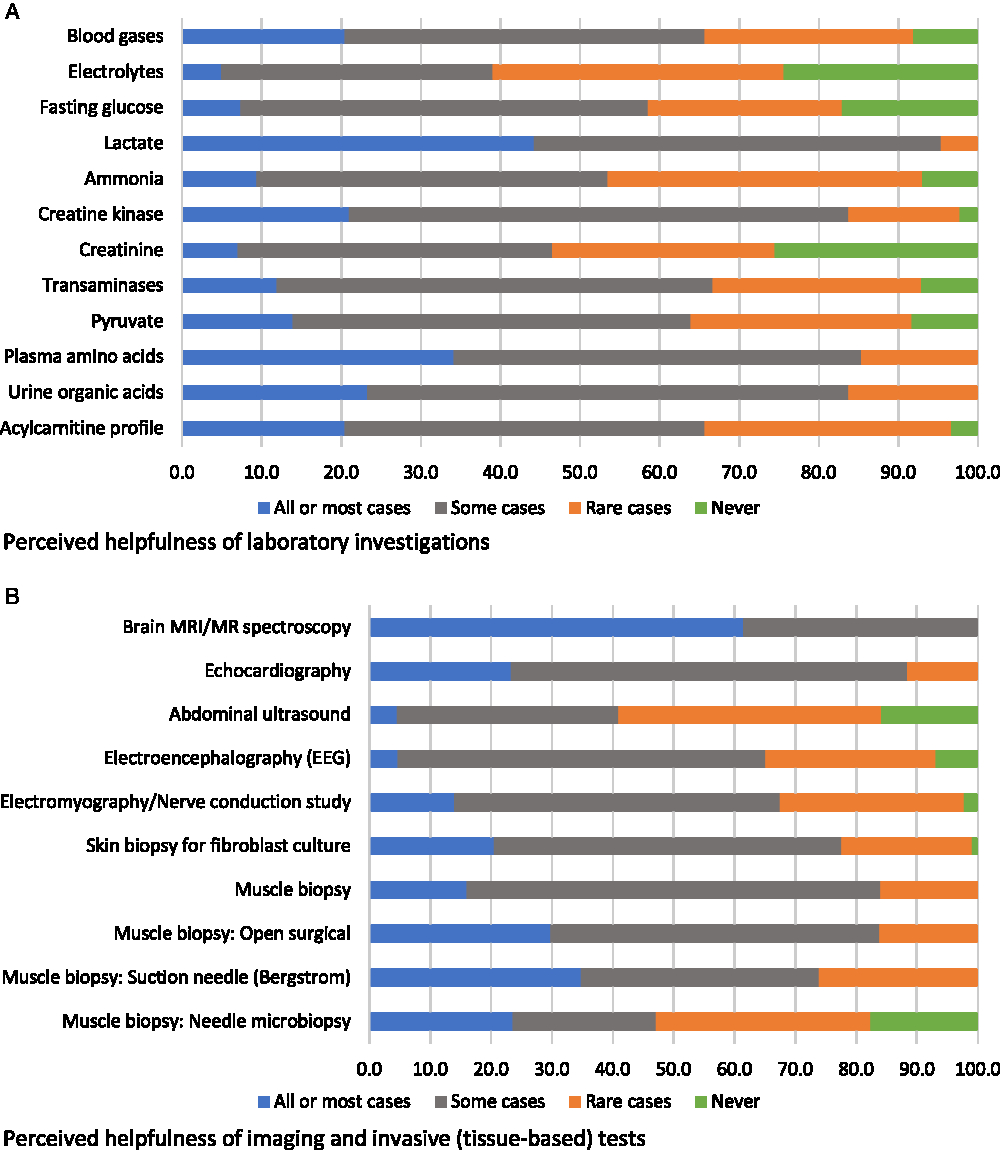
Figure 1: Physicians’ responses regarding the perceived helpfulness of non-genetic diagnostic assessments in the investigation for a suspected mitochondrial disorder (percent of respondents).
A majority of participants (75%) reported using genetic/genomic testing as part of the diagnostic process in 75% or more of their patients with suspected mitochondrial disorders. When asked to estimate the proportion of patients for whom genetic/genomic testing was used as a first-line diagnostic investigation, responses were variable: 28% of respondents reported using genetic testing ahead of any other diagnostic investigations in fewer than 25% of their patients, 14% reported using it for 25%–49% of their patients, 28% reported using it for 50%–74% of their patients, and 30% reported using it for 75% or more of their patients. Despite the wide use of genetic/genomic testing, most respondents (79%) reported having identified the causative mutation in fewer than 50% of their patients. There seemed to be a greater degree of consensus among respondents concerning the perceived helpfulness of various genetic/genomic assessments relative to non-genetic diagnostic assessments: when 20% or more of respondents endorsed a genetic/genomic assessment as helpful in all or most cases, few reported it to be helpful rarely or never (Figure 2). The most highly endorsed genetic/genomic assessments, which were reported as helpful in all or most cases by 36% and 33% of respondents, respectively, were next-generation sequencing (NGS) of the entire mitochondrial genome, and mtDNA testing using muscle tissue. Only one assessment, single gene tests, was indicated by a majority of respondents (51%) to be rarely or never helpful. No genetic/genomic tests were reported as “potentially helpful but not available” by more than 20% of respondents (not shown in figure). The two genetic/genomic assessments most commonly reported as unavailable were whole exome sequencing (16%) and mtDNA testing using urinary sediment (14%). Respondents who reported a lack of availability of urinary sediment mtDNA testing were from Ontario and British Columbia, while those who reported poor availability of whole exome sequencing were from Alberta, Manitoba, and Quebec.
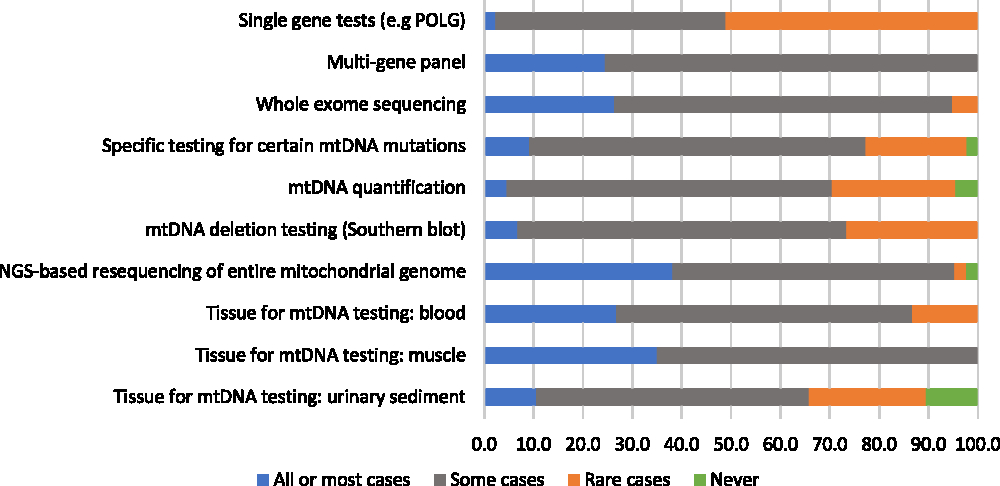
Figure 2: Physicians’ responses regarding the perceived helpfulness of genetic diagnostic assessments in the investigation for a suspected mitochondrial disorder (percent of respondents).
Respondents were also asked generally about the availability of diagnostic methods and their coverage by provincial health insurance. At least 50% of respondents in all Canadian regions (the west coast, the Prairie Provinces, central Canada, and the Atlantic region) reported challenges associated with the availability and/or coverage of the diagnostic investigations they would like to use. A frequently reported challenge, according to survey respondents, was a lack of provincial funding, creating barriers to accessing tests such as whole exome sequencing, NGS, gene panels, and muscle biopsies. Other challenges included the lengthy process to request funding, or that testing must be sent out of the province or the country; this latter barrier was mentioned by nine respondents. Concerns about accessing whole exome sequencing in particular were shared by 11 respondents.
About a quarter (26%) of physicians who participated in the survey also reported that 50% or more of their patients with suspected mitochondrial disorders were in a later stage of disease progression at their first encounter. When asked what they considered to be the single most significant barrier to the clinical recognition of mitochondrial diseases in patients presenting with compatible symptoms, 71% chose non-specific symptoms. Most of the remaining participants (20%) chose the broad differential diagnosis. As the most important barrier to the specific genetic diagnosis of a patient’s mitochondrial disorder, most respondents chose non-specific symptoms (29%), the broad differential diagnosis (27%), or poor availability of molecular diagnostic investigations (24%). As open-ended responses, other participants noted barriers related to disorders that may be missed by gene panels or whole exome sequencing; the scope and invasiveness of investigations; the use of less robust investigations which may provide confusing results; and turnaround time for whole exome sequencing.
Patient Monitoring and Organization of Care
Most participants reported having provided clinical care for patients with mitochondrial disorders within the past 5 years (87%). These respondents were asked to evaluate various monitoring evaluations for mitochondrial disorders according to whether they would use an evaluation routinely “for all or most patients,” “for some patients,” “in rare cases,” or “never.” The most highly endorsed evaluations, which the greatest proportions of respondents said they would use routinely for all or most of patients, were growth parameters (83%), lactate (78%), “in clinic” developmental assessment (75%), plasma amino acids (66%), and periodic echocardiography (66%) (Figure 3). There were two evaluations that a relatively high (but minority) proportion of respondents stated they would rarely or never use: plasma 3-methylglutaconic acid tests (49%) and formal neuropsychological assessment (48%). Six monitoring evaluations met our criterion for reflecting “high variability in physician endorsement”: electroencephalography, Holter monitor, HbA1c, creatinine clearance, acylcarnitine profile, and free/total carnitine (Figure 3).
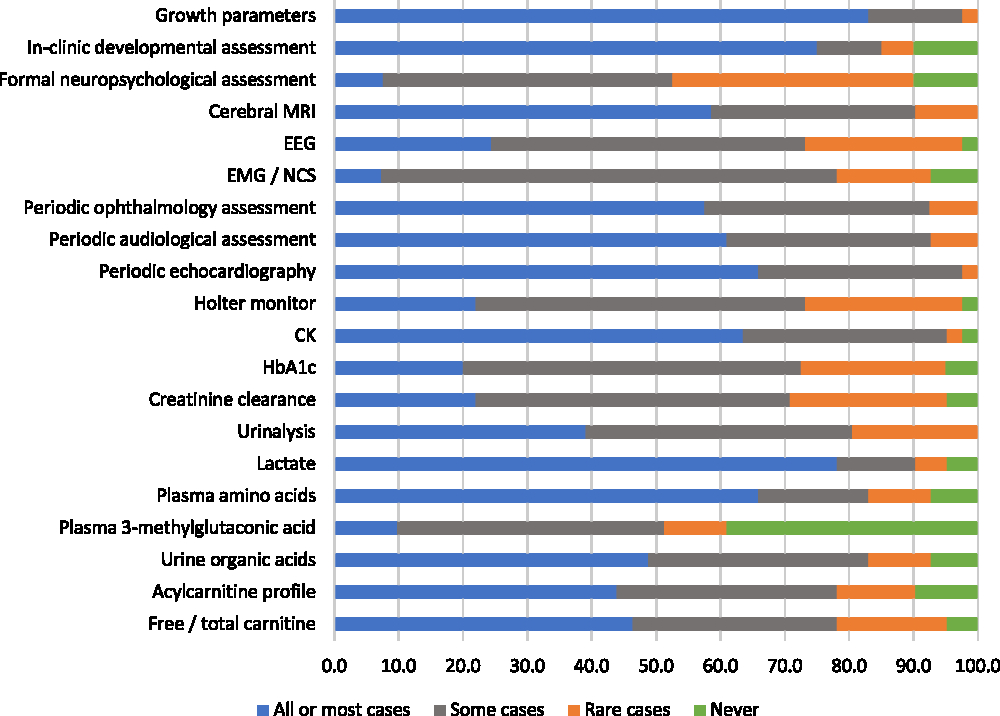
Figure 3: Physicians’ responses regarding the perceived helpfulness of evaluations used to routinely monitor patients diagnosed with mitochondrial disorders (percent of respondents).
Respondents were also asked about the coordination of routine care of patients diagnosed with mitochondrial disorders in their clinical practices. Seventy-one percentage of respondents, who were from all of the regions represented in this survey, reported following any form of an inter-professional care model. When asked to further elaborate on the models in use at their clinics, respondents described collaborating with providers of various disciplines, such as nurse practitioners, dietitians, genetic counsellors, physiotherapists, occupational therapists, psychologists, and social workers. Some respondents described collaborating with other physicians of various specialties within their clinics as well, listing geneticists, pediatricians, neurologists, respirologists, and orthopedic surgeons. In contrast to the large proportion of respondents who indicted the use of an inter-professional model of care, just 40% of respondents, who were exclusively from central Canada or the Prairie Provinces, reported having a care coordination team at their center to which they can refer patients with mitochondrial disorders.
Treatment Practices
Respondents were asked whether they recommend mitochondrial cocktails and single vitamins/cofactors for patients with mitochondrial disorders, for “all or most patients,” “some patients,” “in rare cases,” or “never.” Responses were variable: while 49% would recommend cocktails for all or most patients, 17% would recommend cocktails in rare cases or never (Figure 4). When those who recommend cocktails more than “never” were asked to describe the three mitochondrial cocktails that they recommend most frequently, respondents listed a wide variety of combinations that typically included three to five of the following components (some components were reported for only certain disease-specific cocktails): coenzyme Q10 (ubiquinone), creatine, alpha-lipoic acid, ascorbic acid, vitamin E, thiamin, riboflavin, arginine, and citrulline. The most highly endorsed single vitamins/cofactors, which greater than 30% of respondents said they would recommend for all or most patients, were ubiquinone, riboflavin, alpha-lipoic acid, and carnitine (Figure 4). Four single vitamins/cofactors met our criterion for “high variability in physician endorsement”: creatine, carnitine, alpha-lipoic acid, and ascorbic acid.
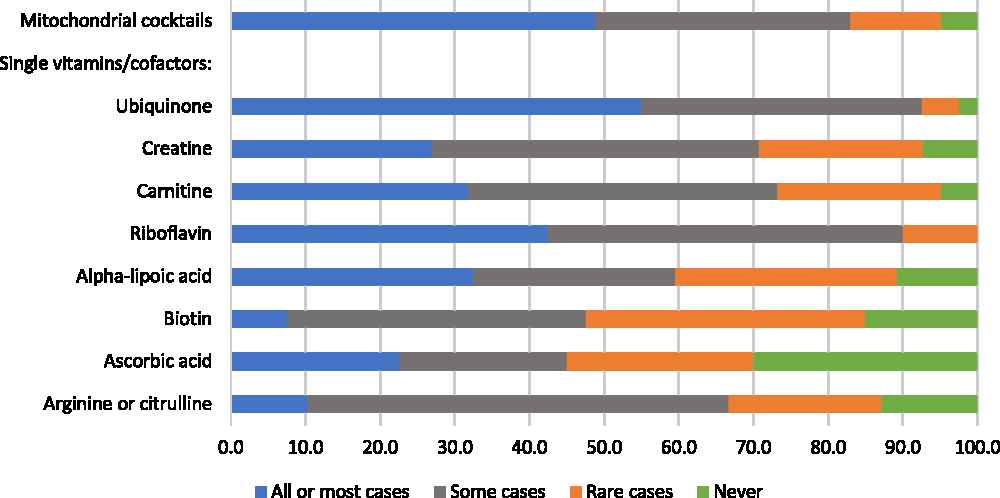
Figure 4: Physicians’ responses regarding the perceived helpfulness of mitochondrial cocktails and single vitamins/cofactors (as part of a cocktail, or separately) for the treatment of patients with mitochondrial disorders (percent of respondents).
Respondents reported that their patients typically access most of the compounds in the mitochondrial cocktails they use as over-the-counter supplements (i.e., non-prescription products), or as licensed drugs. When asked about the availability and coverage of cocktails and/or their components, participants’ responses were variable: 25% reported that all or most of the cocktails they use are available to patients and covered by public insurance. A further 20% reported challenges associated with availability, 28% reported challenges associated with coverage, and 25% reported challenges associated with both availability and coverage of mitochondrial cocktails or their components. Respondents who reported challenges in access to mitochondrial cocktails were located in all of the provinces represented in this survey.
Regarding other interventions for the management of primary mitochondrial disorders, a majority of respondents stated that they would recommend smoking cessation or prevention (75%), avoidance of potentially mitotoxic drugs (78%), and regular moderate exercise (73%) to all or most of their patients. As examples of potentially mitotoxic drugs for patients to avoid, respondents listed propofol, aminoglycosides, valproic acid, halogenated inhaled anesthetics, statins, aspirin, and corticosteroids.
Research Interests and Priorities
About half (49%) of the respondents reported having been involved as an investigator in research related to mitochondrial disorders in the past 5 years. This research consisted of case reports (n = 17), basic research on mitochondrial function (n = 9), and natural history studies (n = 8), as well as development of diagnostics (n = 7), therapies (n = 3), drug development research (n = 4), and clinical trials (n = 4). When asked about the three research areas that are most important to improve outcomes for patients with mitochondrial disorders as an open-ended question, the most common response, suggested by 26 respondents, was for research to develop effective therapies: 11 respondents listed drug development or research on the efficacy of cocktails, supplements, and vitamins; 10 respondents suggested studies on specificity and efficacy of gene therapies; and 5 respondents suggested studies on the impact of exercise and its therapeutic potential. Other common suggestions included research to improve biomarkers for disease monitoring and diagnosis, and diagnostic markers for specific mitochondrial disorders.
Discussion
We identified variation in practice among Canadian physicians providing diagnostic and management care for suspected and confirmed primary mitochondrial disease. With respect to diagnosis, no specific diagnostic evaluation was endorsed by an overwhelming majority of respondents for use in all or most cases. This is likely in part a consequence of the heterogeneous nature of mitochondrial disorders, with investigations dependent on specific pathologies.Reference Haas, Parikh and Falk21 In cases where participants’ opinions of a diagnostic assessment reflected a relatively higher consensus, they generally aligned with the recent MMS guidelines.Reference Parikh, Goldstein and Koenig12 For example, assessments that were regarded positively by a relatively high proportion of respondents, such as brain MRI/MR spectroscopy, lactate, NGS-based sequencing of the full mitochondrial genome, and using muscle tissue for mtDNA testing, were all recommended in the MMS statement; and the one diagnostic assessment that was seen by the majority of respondents to be never or rarely helpful, single gene tests, was discouraged in the guidelinesReference Parikh, Goldstein and Koenig12 Acylcarnitine analysis, which was noted by the MMS guideline authors to have little empirical support despite its being commonly recommended elsewhere as a diagnostic evaluation,Reference Parikh, Goldstein and Koenig12 met our criterion for high variability in physician endorsement. Our findings thus suggest that Canadian mitochondrial physicians have views that generally align with established recommendations but that there remains a need for further evidence to better support decision-making about diagnostic care. This need for evidence is reinforced by our finding that nearly 80% of participants estimated that they identify the mutations responsible for mitochondrial disease in only a minority of their patients.
Generating evidence to establish the clinical validity of diagnostic tests and test strategies for mitochondrial disease is challenging due to both the rarity of specific diseases or syndromes and the absence in many cases of a reference standard that can be used to evaluate emerging tests. Future evaluative studies should follow established principles for assessing diagnostic test accuracy wherever possible, for example, using prospective study designs that enroll unselected patients (e.g., random or sequential patients suspected of a mitochondrial disease) and ensuring comparable follow-up.Reference Cohen, Korevaar and Altman22, Reference Whiting, Rutjes and Westwood23 Stratification by phenotype may be important as the value of particular diagnostic strategies may differ depending on clinical manifestations and patient characteristics. In addition to evaluating the clinical validity of diagnostic tests using metrics such as sensitivity, specificity, and diagnostic utility (ability of a testing strategy to eventually lead to an etiologic diagnosis), future studies could also evaluate which sequence of ordering of tests leads to the most timely diagnoses for particular presenting characteristics. Studies should also investigate the clinical utility of diagnostic approaches with respect to associations between changes to diagnostic care and changes to disease management and/or patient and family-centered outcomes.24
Some survey respondents reported challenges associated with the availability or coverage of diagnostic assessments, with 24% of respondents identifying poor availability of molecular diagnostic investigations as the most important barrier to patients receiving a specific genetic diagnosis. Despite these perceived barriers, we note that a full range of diagnostic services are currently available with cost coverage through major Canadian academic health sciences centers that have specialist expertise in mitochondrial disorders. It may be that perceived barriers related to the access and availability of some tests, including administrative barriers that result in delays to accessing tests, differ depending on the size or other characteristics of specific centers. However, we were unable to investigate perceptions of diagnostic test availability within specific cities or hospitals due to our small sample size. We note that 49% of respondents were from Ontario, where mtDNA sequencing by NGS as well as NGS for nuclear genes affecting the mitochondria are currently available and provincially supported.
Challenges related to the non-specific symptoms and broad differential diagnosis were cited as the most important barriers to achieving a specific genetic diagnosis of mitochondrial disease by 29% and 27% of respondents, respectively. In addition, more than one quarter of participating physicians reported that a majority of their patients with suspected mitochondrial disorders were in a later stage of disease progression at their first encounter. Collectively, these findings regarding the views of physicians about the challenges in diagnosing mitochondrial disorders corroborate the results of a recent survey describing the “diagnostic odyssey” of 215 patients or family members of patientsReference Grier, Hirano, Karaa, Shepard and Thompson25; in that study, patients consulted a median of five physicians during the diagnostic process and a majority had received at least one non-mitochondrial diagnosis before being diagnosed with mitochondrial disease. Prospective patient-level data, for example from a representative cohort, documenting the time from the onset of first symptoms to care-seeking contact, and to the final diagnosis, is currently unavailable but would be helpful for further elucidating the diagnostic journey and evaluating changes to diagnostic care.
In addition to the diagnostic odyssey, patients with mitochondrial disorders frequently face challenges related to their high needs for health services across multiple medical specialties, with coordinated multidisciplinary care recognized as important from the perspectives of patients/familiesReference Anderson, Elliott and Zurynski26 and providers.Reference Parikh, Goldstein and Karaa13 While a majority of physicians participating in our survey reported following an inter-professional model of care (71%), only 40% perceived an ability to refer patients with mitochondrial disorders to a care coordination team. This may be a particularly important barrier for patients and their families given our finding that most physicians spend a limited amount of their clinical time caring for patients with mitochondrial disease; many individual institutions may thus not have extensive and centralized expertise about the needs of this rare disease population. According to a recently published online catalogue, at least 11 tertiary care pediatric centers in Canada have some form of complex care service provision available although not all involve complex care clinics or coordinators and criteria for accessing services vary.27 Further research is needed to better understand the experiences with care of patients with mitochondrial disorders and their families, including their needs for and access to coordination services as well as their experiences with diagnostic care and perceptions about priority gaps in care.
With respect to disease management, just under half (49%) of physicians participating in our survey reported that they would recommend mitochondrial cocktails for all or most patients, with variation in responses regarding specific vitamins and cofactors as either ingredients in cocktails or as single interventions. While acknowledging a 2012 Cochrane review that identified no strong evidence in support of the efficacy of vitamins and cofactors,Reference Pfeffer, Majamaa, Turnbull, Thorburn and Chinnery6 the MMS consensus guidelines recommend offering coenzyme Q10/ubiquinone, alpha-lipoic acid, and riboflavin to all individuals with mitochondrial disorders,Reference Parikh, Goldstein and Koenig12 corresponding to the three most highly endorsed single vitamins/cofactors among our survey respondents. Nearly three quarters (73%) of respondents also noted that they would recommend regular moderate exercise to all or most individuals with mitochondrial disorders, consistent with the guidelines.Reference Parikh, Goldstein and Koenig12 Thus, as with diagnostic testing, Canadian physicians’ views on treatment for mitochondrial disease are generally aligned with established recommendations. Our survey respondents also recognized the absence of strong evidence in support of all therapies for mitochondrial disease, with a majority of participants citing studies on the development of effective therapies as the top research priority in the field. Notably, 20% of survey respondents reported challenges associated with availability, 28% reported challenges associated with coverage, and 25% reported challenges associated with both availability and coverage of mitochondrial cocktails or their components.
This study is the first survey of Canadian physicians who diagnose and manage primary mitochondrial disorders and contributes important information about the organization of care and use of specific diagnostic and treatment strategies. However, the findings should be interpreted in light of some limitations. For example, our non-random sampling strategy and non-response of close to half of sampled physicians may have created selection bias so that our findings may not represent all Canadian physicians involved in care for mitochondrial disorders. We did not collect information about non-respondents, rendering it challenging to evaluate this possibility. A related issue was our small sample size, which meant we were unable to investigate systematic differences in responses according to physician specialty; views from specialties other than medical genetics and general pediatrics (which made up the majority of specialties in our participant population) may not be well reflected in our findings. Furthermore, it is possible that different respondents interpreted the questions differently, particularly when asked to indicate how helpful different interventions were likely to be, given the inherently subjective nature of the response choices. While this question format was necessary due to the heterogeneity of mitochondrial disorders, our a priori definition of high variation in physician endorsement allowed us to identify key areas of uncertainty. Finally, results from this survey provide only a partial perspective regarding the nature of challenges associated with the diagnosis and ongoing care of mitochondrial disorders. Studies that seek to understand the patient and family perspective on these issues would add important information to the results reported here, particularly given that the aspects of disease and care that are prioritized by patients may differ from those prioritized by clinicians.Reference Koene, Wortmann and de Vries28
Conclusions
Perhaps due to the relatively low incidence of each type of disorder, the nature of care and variation in care provided for individuals with mitochondrial disorders in Canada has not been well documented to date. Our findings indicate that while Canadian physicians’ views about diagnostic care and disease management are aligned with published recommendations, there are important variations in care that reflect persistent areas of uncertainty. In order to support the development and updating of standard protocols and encourage best practices, our results underscore the need for well-designed investigations of the diagnostic accuracy of biochemical, imaging, and genetic-genomic assessments and of the clinical efficacy of various treatments, including mitochondrial cocktails and their components for this large group of rare diseases. Future research should also incorporate the patient and family perspective regarding experiences with both diagnostic care and ongoing management, with particular attention to care coordination needs.
Acknowledgements
This study was funded through a Canadian Institutes of Health Research Emerging Team Grant (TR3-119195). This study was conducted in collaboration with the Canadian Inherited Metabolic Diseases Research Network, working in partnership with MitoCanada. We are grateful for the contributions of the survey participants.
Disclosures
MT is a major shareholder, president and CEO of Exerkine Corporation, which has products in development for the treatment of mitochondrial disease/dysfunction; and has received speaker honoraria from Sanofi-Genzyme. KS is a local site Principal Investigator for a study funded by Biomarin that is not related to mitochondrial disorders. SD reports personal fees from Shire, personal fees and non-financial support from Biomarin, and non-financial support from Genzyme, outside the submitted work. The other authors have no conflicts of interest to disclose related to this paper.
Statement Of Authorship
KP contributed to the design of the study, the analysis and interpretation of the results, and drafting the manuscript. MAL, PC, SDK, and ML contributed to the conception and design of the study, the interpretation of the results, and revisions to the manuscript for important intellectual content. KT contributed to the analysis and interpretation of the results and revisions to the manuscript for important intellectual content. WA, CBG, AK, BP, CRG, CAR, NS, DC, SD, AF, MTG, JG, CDMVK, JBK, JL, MP, KS, RS, LAT, KW, and DB contributed to the design of the study, the interpretation of the results, and revisions to the manuscript for important intellectual content. BKP contributed to the conception, design, and supervision of the study, the interpretation of the results, and drafting the manuscript. All authors approved the final submitted version of the manuscript.






