


Astaxanthin (3,3′-dihydroxy-β,β′-carotene-4,4′-dione) is a lipophilic pigment with a reddish colour, which is naturally synthesised by algae or plants, and is primarily distributed in marine organisms. For example, salmon, shrimp, and crab are representative astaxanthin-containing seafood and their red colour is related to astaxanthin. Also, astaxanthin is commercially available as a nutritional supplement in many countries including the USA, Sweden, Japan and South Korea. Astaxanthin belongs to the xanthophyll group, which are oxygenated derivatives of carotenes. The xanthophylls and carotenes are two major groups of carotenoids that possess a number of health benefits. Like other carotenoids, astaxanthin has antioxidant, anti-cancer and anti-inflammatory activities(Reference Higuera-Ciapara, Félix-Valenzuela and Goyocoolea1). However, astaxanthin's most beneficial property is its activity as a potent antioxidant. Miki(Reference Miki2) reported that the antioxidant activity of astaxanthin was more powerful than that of β-carotene or α-tocopherol, well-known antioxidants. Moreover, the biological activities of astaxanthin have been examined in clinical studies regarding CVD, male infertility, functional dyspepsia and age-related maculopathy(Reference Pashkow, Watumull and Campbell3–Reference Parisi, Tedeschi and Gallinaro6).
In humans, the bioavailability of carotenoids is low and variable. It ranges from 10 to 50 % of a given dose, due to low solubility in gastrointestinal tract juices, leading to poor absorption by the epithelial cells of the small intestine(Reference Nagao7). It has been reported that the low bioavailability of astaxanthin is improved by lipid-based formulations in human subjects(Reference Odeberg, Lignell and Pettersson8). Also, the oral bioavailability of astaxanthin could be improved by timing the dose to be taken either before or after a meal(Reference Okada, Ishikura and Maoka9). Showalter et al. (Reference Showalter, Weinman and Østerlie10) found in mice that after both single and multiple oral administrations, the total area under the concentration–time curve (AUC) of astaxanthin in the liver was greater than that in the plasma, suggesting considerable uptake by the liver. Petri & Lundebye(Reference Petri and Lundebye11) evaluated the tissue distribution of astaxanthin after oral administration for 2 weeks in rats and found that astaxanthin accumulated primarily in the spleen, kidneys, adrenals, liver, skin and eyes. Recently, several studies reported the metabolism of astaxanthin in animals and human subjects. On the basis of in vivo human (per oral administration) and in vitro human hepatocyte studies, astaxanthin is metabolised into 3-hydroxy-4-oxo-β-ionol and 3-hydroxy-4-β-ionone(Reference Kistler, Liechti and Pichard12). On the basis of in vitro primary cultures of rat hepatocytes, astaxanthin is metabolised to 3-hydroxy-4-oxo-β-ionone and its reduced form, 3-hydroxy-4-oxo-7,8-dihydro-β-ionone, and their glucuronide conjugates(Reference Erich, Hans and Brightte13). However, the biological effects of the metabolites of astaxanthin have not been thoroughly studied. Also, the pharmacokinetics and first-pass extraction of astaxanthin were not reported.
The objective of the present study was to determine the pharmacokinetics of astaxanthin after its intravenous (at doses of 5, 10 and 20 mg/kg) and oral (at doses of 100 and 200 mg/kg) administration, and its gastrointestinal and hepatic first-pass extraction ratios, following intravenous, intragastric or intraportal administration at a dose of 20 mg/kg in rats. Hepatic cytochrome P-450 1A1/2, an enzyme responsible for the metabolism of astaxanthin, and its tissue distribution in rats were also examined.
Materials and methods
Chemicals
Astaxanthin, β-apo-8-carotenal (internal standard for the analysis of astaxanthin by HPLC), 3-methylcholanthrene (inducer of cytochrome P-450 1A1/2 in rats(Reference Correia14)), maize oil and N,N-dimethylacetamide were purchased from Sigma-Aldrich Corporation (St Louis, MO, USA). Polyethylene glycol 400 was from Showa Chemical Company (Tokyo, Japan). Other chemicals were of reagent or HPLC grade (Mallinckrodt Baker, Inc., Phillipsburg, NJ, USA).
Animals
The protocol for the present animal study was approved by the Institute of Laboratory Animal Resources of Seoul National University, Seoul, South Korea.
Male Sprague–Dawley rats (7–8 weeks old, weighing 240–330 g) were purchased from Charles River Company Korea (Orient, Seoul, South Korea). Rats were maintained in a light-controlled room (light: 7.00–19.00 hours, dark: 19.00–7.00 hours), kept at a temperature of 22 ± 2°C and a relative humidity of 55 ± 5 % (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University). Rats were housed in metabolism cages (Tecniplast, Varese, Italy) maintained with a ad libitum supply of filtered pathogen-free air, food (Agribrands Purina Korea, Pyeongtaek, South Korea) and water.
Intravenous and oral studies
The procedures used for the pretreatment of rats including the cannulation of the carotid artery and the jugular vein were similar to a previously reported method(Reference Kim, Choi and Lee15). Cannulation of the carotid artery for blood sampling was done early in the morning and all intravenous drugs were delivered through the access in the jugular vein. Astaxanthin (dissolved in polyethylene glycol 400–N,N-dimethylacetamide (50:50, v/v) at doses of 5 (n 8), 10 (n 8) and 20 (n 6) mg (2 ml)/kg was manually infused for 1 min via the jugular vein. Blood samples, approximately 0·22 ml each, were collected via the carotid artery at 0 (control), 1 (end of the infusion), 5, 15, 30, 60, 90, 120, 240, 360, 480, 720, 1200 (5 mg/kg) and 1440 min (10 and 20 mg/kg) after the start of the intravenous infusion of astaxanthin. Heparinised 0·9 % NaCl-injectable solution (20 units/ml; 0·3 ml) was used to flush the cannula after each blood sampling to prevent blood clotting. Each blood sample was immediately centrifuged and 100 μl of each plasma sample was stored at − 70°C until its use in the HPLC analysis of astaxanthin. The procedures used for the preparation and handling of the 24 h urine sample were similar to a previously reported method(Reference Kim, Choi and Lee15). Astaxanthin, using the same solution as used in the intravenous study, at doses of 100 (n 8) and 200 (n 9) mg (5 ml)/kg was administered orally using a gastric gavage to rats. Blood sampling time schedules were 0, 2, 4, 6, 7, 8, 12, 24, 30, 36 (100 mg/kg) and 48 h (200 mg/kg) after the oral administration of astaxanthin. Other procedures for the oral study were similar to those of the intravenous study. In the present oral studies, higher oral doses than in human subjects (40 mg) were used to obtain a complete and more reliable pharmacokinetic parameters because the gastrointestinal absorption of astaxanthin is low.
Measurement of first-pass extraction ratio of astaxanthin in rats
The procedures used were similar to the previously reported methods(Reference Murakami, Nakanishi and Yoshimori16, Reference Choi, Kim and Lee17). The vein from the caecum was cannulated and the cannula was pushed forward about 4·0 cm toward the liver through the portal vein to minimise impaired blood flow into the portal vein(Reference Murakami, Nakanishi and Yoshimori16). For the intravenous administration (n 4), astaxanthin, using the same solution as used in the intravenous study, at a dose of 20 mg (2 ml)/kg was infused for 30 min via the jugular vein. The same volume of the vehicle (polyethylene glycol 400–N,N-dimethylacetamide (50:50, v/v)) was infused for 30 min into the portal vein and instilled into the stomach using a syringe after 4–5 h recovery from the light diethyl ether anaesthesia. All intravenous administration was applied with the assistance of an infusion pump (Model 2400-006; Harvard Instrument, South Natick, MA, USA). For intraportal administration (n 3), the same dose of astaxanthin was infused for 30 min via the portal vein and the same volume of the vehicle was infused for 30 min into the jugular vein and instilled into the stomach. For the intragastric administration (n 4), the same dose of astaxanthin was instilled into the stomach and the same volume of the vehicle was infused for 30 min via the jugular and portal veins. Blood samples (approximately 0·22 ml each) were collected via the carotid artery at 0, 15, 30 (end of the infusion), 35, 45, 60, 90, 150, 270 and 510 min for the infusion study, and at 0, 60, 120, 180, 240, 360 and 480 min for the instillation study.
Administration of 3-methylcholanthrene, a cytochrome P-450 1A1/2 inducer, in rats
Rats received four daily intraperitoneal injections of 20 mg (3·3 ml)/kg of 3-methylcholanthrene group)(Reference Williams, Lowitt and Szentivanyi18, Reference Choi, Kim and Lee19) or 3·3 ml/kg of maize oil (3-methylcholanthrene control rats group). During the pretreatment, rats had free access to food and water. The experiment was performed on day 5 after the start of the pretreatment. Other procedures were similar to those in the previously described intravenous study. T and C refer to the treated and control, respectively.
Tissue distribution of astaxanthin after its oral administration in rats
The procedures used were similar to a previously reported method(Reference Lee, Kim and Kim20). Astaxanthin, the same solution as used in the oral study, at a dose of 100 mg (5 ml)/kg was administered orally using a gastric gavage tube in rats. Approximately, 1 g of each liver, kidney, heart, stomach, lung, small intestine, large intestine, spleen, fat, muscle and brain was quickly excised after 8 and 24 h, respectively (n 3, each) after cervical dislocation. Each tissue sample was rinsed with cold 0·9 % NaCl-injectable solution, blotted dry with tissue paper and weighed. Then, each tissue was homogenised with four volumes of 0·9 % NaCl-injectable solution in a tissue homogeniser (Ultra-Turrax T25; Janke & Kunkel, IKA-Labortechnik, Staufeni, Germany) and centrifuged. Two 50 μl aliquots of plasma samples or the supernatant of each tissue homogenate were stored at − 70°C until use for the HPLC analysis of astaxanthin.
Stability of astaxanthin
The procedures used were similar to a previously reported method(Reference Yu, Bae and Kim21). An astaxanthin stock solution (dissolved in chloroform–hexane (30:70, v/v)) was spiked (10 μl/ml) in each test-tube that contained four rat gastric juices and various buffer solutions with pH ranging from 1 to 13 to produce an astaxanthin concentration of 2 μg/ml. After vortex-mixing, each sample was incubated in a water-bath shaker kept at a temperature of 37·8°C and at a rate of 50 oscillations/min at 0, 15, 30, 60, 120 and 240 min for rat gastric juices and at 2, 4, 6, 8, 12 and 24 h for various buffer solutions. Then, 100 μl were sampled from each test-tube. The concentrations of astaxanthin in the previously mentioned samples were analysed as soon as the sample was collected.
HPLC analysis of astaxanthin
Concentrations of astaxanthin were analysed by a slight modification of the reported HPLC method(Reference Okada, Ishikura and Maoka9). Briefly, 600 μl of acetone containing 2 μg/ml of β-apo-8-carotenal, as an internal standard, was added to 100 μl of a sample. After vortex-mixing for 1 min, the mixture was extracted with 600 μl of hexane. The organic layer was collected and dried (Dry thermobath, Eyela, Tokyo, Japan) under a gentle stream of N2 gas at 25°C. A 100 μl aliquot of the mobile phase was added to reconstitute the residue and 30 μl was directly injected onto a normal-phase HPLC column. The mobile phase, hexane–acetone (75:25, v/v), was run at a flow-rate of 1·0 ml/min, and the column eluent was monitored using a UV detector at 470 nm, at room temperature. The retention times of the internal standard and astaxanthin were approximately 2·3 and 4·8 min, respectively. The detection limits of astaxanthin in rat plasma and urine samples were 0·01 and 0·05 μg/ml, respectively. The CV of astaxanthin in plasma and urine samples were < 9·6 and 7·8 %, respectively.
Pharmacokinetic analysis
Standard methods(Reference Gibaldi and Perrier22) were used to calculate the following pharmacokinetic parameters using a non-compartmental analysis (WinNonlin®; professional edition version 2.1; Pharsight, Mountain View, CA, USA): the AUC(Reference Chiou23), terminal half-life, time-averaged total body, renal and non-renal clearances (CL, CLR and CLNR, respectively), mean residence time and apparent volume of distribution at steady state. The peak plasma concentration (C max) and time to reach C max were directly read from the experimental data. Doses were normalised based on 1 mg/kg of astaxanthin for AUC and C max for statistical analysis.
Statistical analysis
Data were analysed individually and a value of P < 0·05 was deemed to indicate statistical significance. Statistical evaluations were performed using Student's t test between the two means for the unpaired data or Duncan's multiple range test of SPSS (Chicago, IL, USA) a posteriori ANOVA among the three means for the unpaired data. All the data are expressed as mean and standard deviation, except median (range) for time to reach the C max.
Results
Pharmacokinetics of astaxanthin after its intravenous and oral administration in rats
The mean arterial plasma concentration–time profiles for the intravenous administration of astaxanthin at doses of 5, 10 and 20 mg/kg are shown in Fig. 1(a). The relevant pharmacokinetic parameters are listed in Table 1. The dose-normalised AUC of astaxanthin at a dose of 20 mg/kg (61·0 (sd 15·7) μg min/ml) was significantly greater by 54·8 % than that at 5 mg/kg (39·4 (sd 9·90) μg min/ml). Similar results were obtained from CL and CLNR. The CLR at 10 and 20 mg/kg were significantly slower (66·4 and 86·0 %, respectively) than that at 5 mg/kg. These data suggest that astaxanthin exhibits dose-dependent pharmacokinetics after three intravenous doses, as studied in rats. The CL and total amount excreted in 24 h urine were almost negligible.
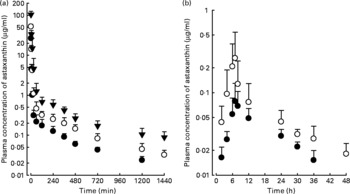
Fig. 1 Mean arterial plasma concentration–time profiles of astaxanthin after its intravenous administration (a) at doses of 5 (●; n 8), 10 (○; n 8), and 20 (▾; n 6) mg/kg and oral administration (b) at doses of 100 (●; n 8) and 200 (○; n 9) mg/kg in rats. Bars indicate standard deviations.
Table 1 Pharmacokinetic parameters of astaxanthin after its intravenous and oral administration at various doses in rats
(Mean values and standard deviations)

AUC, total area under the plasma concentration–time curve from time zero to infinity; MRT, mean residence time; CL, time-averaged total body clearance; CLR, time-averaged renal clearance; CLNR, time-averaged non-renal clearance; V ss, apparent volume of distribution at steady state; C max, peak plasma concentration; T max, time to reach C max; Ae 0–24 h, total amount excreted in 24 h urine; Ae 0–48 h, total amount excreted in 48 h urine.
* AUC and C max values were normalised to the astaxanthin dose of 1 mg/kg for statistical analyses.
† Mean values for 5 mg/kg were significantly different from 20 mg/kg (P < 0·05).
‡ Mean values for 5 mg/kg were significantly different from 10 and 20 mg/kg (P < 0·05).
§ Range of T max.
The mean arterial plasma concentration–time profiles for the oral administration of astaxanthin at doses of 100 and 200 mg/kg are shown in Fig. 1(b). The relevant pharmacokinetic parameters are also listed in Table 1. The dose-normalised AUC of astaxanthin at a dose of 100 mg/kg (0·773 (sd 0·162) μg min/ml) was comparable to that at a dose of 200 mg/kg (0·750 (sd 0·385) μg min/ml). The dose-normalised C max of astaxanthin at a dose of 200 mg/kg (1·69 (sd 1·34) ng/ml) was higher than the C max at a dose of 100 mg/kg (0·956 (sd 0·456) ng/ml), but not statistically significant. Similar results were also obtained for other pharmacokinetic parameters. The previously mentioned data suggest that astaxanthin exhibits dose-independent pharmacokinetics after two oral doses of astaxanthin in rats.
Hepatic and gastric first-pass extraction ratios of astaxanthin in rats
Following the intravenous, intraportal or intragastric administration of astaxanthin at a dose of 20 mg/kg, its mean arterial plasma concentration–time profiles of astaxanthin were determined, as shown in Fig. 2. The AUC of astaxanthin after its intraportal administration (307 (sd 14·8) μg min/ml) was significantly smaller (by 49·0 %) than after its intravenous administration (602 (sd 69·3) μg min/ml), suggesting that the hepatic first-pass extraction ratio of astaxanthin after absorption into the portal vein was approximately 0·49 in rats.

Fig. 2 Mean arterial plasma concentration–time profiles of astaxanthin after its intravenous (●; n 4), intraportal (○; n 3) and intragastric (▲; n 4) administration at a dose of 20 mg/kg in rats. Bars indicate standard deviations.
The AUC of astaxanthin after its intragastric administration (30·5 (sd 4·41) μg min/ml) was significantly smaller, by 90·1 %, than after intraportal administration. The previously mentioned data suggest that the gastrointestinal first-pass extraction ratio of astaxanthin was approximately 0·901 of the dose in rats.
Pharmacokinetics of astaxanthin in rats pretreated with 3-methylcholanthrene
The mean arterial plasma concentration–time profiles of astaxanthin after its intravenous administration in rats pretreated with 3-methylcholanthrene and control rats are shown in Fig. 3. Relevant pharmacokinetic parameters are listed in Table 2. Compared with 3-methylcholanthrene control rats rats, the AUC was significantly smaller, by 26·6 %, mean residence time was significantly shorter, by 16·2 %, and CL and CLNR were significantly faster, both by 30·1 %, in pretreated with 3-methylcholanthrene rats. The previously mentioned data suggest that astaxanthin is metabolised primarily by cytochrome P-450 1A1/2 in rats.
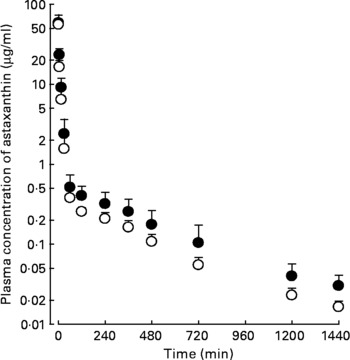
Fig. 3 Mean arterial plasma concentration–time profiles of astaxanthin after its intravenous administration at a dose of 20 mg/kg in rats pretreated with 3-methylcholanthrene (○; n 8) and without 3-methylcholanthrene (●; n 8). Bars indicate standard deviations.
Table 2 Pharmacokinetic parameters of astaxanthin after its intravenous administration at a dose of 10 mg/kg in rats pretreated with 3-methylcholanthrene (MC-T) and without 3-methylcholanthrene (MC-C)
(Mean values and standard deviations)

AUC, total area under the plasma concentration–time curve from time zero to infinity; MRT, mean residence time; CL, time-averaged total body clearance; CLR, time-averaged renal clearance; CLNR, time-averaged non-renal clearance; V ss, apparent volume of distribution at steady state; Ae 0–24 h, total amount excreted in 24 h urine.
* Mean values for MC-T were significantly different from MC-C (P < 0·05).
Tissue distribution of astaxanthin after its oral administration in rats
The amounts of astaxanthin recovered from each tissue at 8- and 24 h after its oral administration at a dose of 100 mg/kg are listed in Table 3. Tissue:plasma ratios of astaxanthin at 8 and 24 h were greater than unity for all tissues studied, except for the heart at 8 h. This suggests that rat tissues and organs had a high affinity for astaxanthin and this was supported by the large values of volume of distribution at steady state, 4860–5770 ml/kg (Table 1).
Table 3 Amount of astaxanthin recovered from each gram tissue (μg/ml for plasma or μg/g for other tissues) 8 and 24 h after its oral administration at a dose of 100 mg/kg in rats
(Mean values and standard deviations)

T/P ratio, tissue-to-plasma ratios.
Stability of astaxanthin
Astaxanthin was unstable in rat gastric juices: 45·8, 23·1, 49·2 and 56·8 % of the spiked amounts of astaxanthin were recovered after 4 h incubation in four rat gastric juices having pH of 3·0, 3·5, 4·0 and 4·0, respectively. Astaxanthin was also unstable up to 24 h incubation in various buffer solutions: 7·1, 4·8, 22·8, 12·6, 11·0, 12·6, 4·5, 6·7, 5·1, 4·8, 6·0, 11·8 and 9·3 % of the spiked amounts of astaxanthin were recovered in buffer solutions with pH 1–13, respectively. These data suggest that astaxanthin is unstable with regard to chemical and enzymatic degradation.
Discussion
After the intravenous administration of astaxanthin at doses of 5, 10 and 20 mg/kg in rats, the total amount excreted in 24 h urine values of astaxanthin were < 0·301 % of the three intravenous doses studied and the contribution of CLR to CL of astaxanthin was almost negligible (Table 1). This suggests that most of the intravenously administrated astaxanthin is eliminated via a non-renal route (CLNR).
After the intravenous administration of astaxanthin, the dose-normalised AUC at 20 mg/kg was significantly greater than that at 5 mg/kg (Table 1). This could be due to saturation of hepatic metabolism of astaxanthin. This was supported by the significantly slower CLNR of astaxanthin at 20 mg/kg than at 5 mg/kg. However, after the oral administration of astaxanthin, the AUC were linear between 100 and 200 mg/kg (Table 1). After the intravenous administration of astaxanthin, the AUC were also linear between 5 and 10 mg/kg (Table 1). The AUC of intravenous astaxanthin at 5 and 10 mg/kg (in the linear ranges) were 197 and 488 μg min/ml, respectively (Table 1). The AUC of oral astaxanthin up to 200 mg/kg was 150 μg min/ml, smaller than the minimum of 197 μg min/ml obtained for intravenous condition. Thus, linear AUC after oral administration were expected.
After the oral administration of astaxanthin, its terminal half-lives at 1010–1460 min were considerably longer than the half-lives at 470–569 min after its intravenous administration (Table 1). This difference could be caused by an absorption pattern of astaxanthin that follows the flip-flop model. When absorption is considerably slower than elimination, that is, if the absorption rate constant is slower than the elimination rate constant, a ‘flip-flop’ system exists(Reference Boxenbaum24). The half-lives of astaxanthin during the absorption phase after the oral administration of astaxanthin were estimated using a residual method; the estimated values were 528 and 550 min for the astaxanthin oral doses of 100 and 200 mg/kg, respectively, which were close to the half-lives after its intravenous administration (Table 1).
In the present study, the plasma concentrations of 3-hydroxy-4-oxo-β-ionone and 3-hydroxy-4-oxo-7,8-dihydro-β-ionone, the metabolites of astaxanthin, were not measured. Their biological activities were not reported in previous studies. Goto et al. (Reference Goto, Kogure and Abe25) suggested that the potent antioxidant activity of astaxanthin was due to its unique structure at the terminal ring moiety. Thus, it could expected that the metabolites of astaxanthin would not seem to be active as antioxidants(Reference Goto, Kogure and Abe25).
The AUC of astaxanthin after its oral administration at a dose of 40 mg in human subjects, 80·8 μg min/ml(Reference Odeberg, Lignell and Pettersson8), was close to the AUC of 77·3 μg min/ml after its oral administration at a dose of 100 mg/kg in rats (Table 1). This suggests that the oral doses of astaxanthin in rats (100 mg/kg) and human subjects (40 mg) have similar AUC values. The parenteral administration of astaxanthin in clinical research has not been allowed because of its lipophilicity.
Interestingly, the pharmacokinetics of β-carotene, another carotenoid, is affected by co-administration with other nutrients or drugs. When β-carotene was given during administration of orlistat, a lipase inhibitor, the absorption of β-carotene decreased significantly(Reference Zhi, Melia and Koss-Twardy26). On the other hand, co-administration of β-carotene resulted in significant reductions in plasma concentrations of α-tocopherol or canthaxanthin(Reference Xu, Plezia and Alberts27, Reference White, Stacewicz-Sapuntzakis and Erdman28). However, limited information has been reported about drug or food interactions of astaxanthin and further studies are needed to clarify the interactions of astaxanthin.
In summary, after the intravenous (5, 10 and 20 mg/kg) and oral (100 and 200 mg/kg) administration of astaxanthin, its pharmacokinetic parameters were dose dependent and dose independent, respectively, in rats. The absorption of astaxanthin after oral administration followed the flip-flop model. The hepatic and gastrointestinal first-pass extraction ratios were approximately 0·490 and 0·901, respectively. Astaxanthin was unstable up to a 4 h incubation in the rat gastric juices and 24 h incubation in various buffer solutions having pH of 1–13.
Acknowledgements
The present work was supported by a grant of the Korea Health 21 R&D project, Ministry of Health and Welfare, Korea (A050376). The contributions of the authors to the manuscript are as follows: H. D. C. designed and conducted the study, analysed the data and wrote the paper; H. E. K. and S. H. Y. conducted the study; M. G. L. and W. G. S. designed the study and reviewed the manuscript. All the authors read and approved the final manuscript. There are no conflicts of interest to declare.






