


Imprinting or programming as a result of early life experience is becoming an accepted scientific phenomenon. Implicit in this is the concept of a ‘stage of developmental plasticity, where specific conditions give rise to later life outcomes’. The most significant aspect is metabolic imprinting, in which maternal undernutrition, obesity and diabetes during gestation and lactation can contribute towards obesity in the offspring(Reference Levin1). Other endpoints that seem to be affected by early life exposure include neurodevelopment and immune modulation.
The concept of fetal growth affecting adult disease was explained by Barker(Reference Barker2, Reference Barker3) in his seminal papers. Programming later evolved to mean alterations in nutrition and growth at specific developmental points, resulting in long-term or even permanent effects(Reference Lucas4).
Observation is the first step and the initial link between health and early diet is often found from epidemiological investigations. One of the most exemplary studies is the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort(Reference Ness5). This has been the source for a host of publications in which early diet is linked to later obesity and to other health endpoints, as outlined in Table 1.
Table 1 Correlations between early exposure and later health outcomes from the Avon longitudinal study of parents and children cohort

Other cohorts include the Helsinki study, which showed a link between prenatal and postnatal factors and type 2 diabetes(Reference Eriksson, Forsén and Tuomilehto6), diet in pregnancy and blood pressure of offspring(Reference Campbell, Hall and Barker7), maternal nutritional status and blood pressure(Reference Godfrey, Forrester and Barker8) and the USA National Children's Study. This latter study is designed to examine the effects of environmental influences on the health and development of 100 000 children across the USA, following them from before birth until age 21. The goal of the study is to improve the health and well-being of children by assessing the impact of exposure on health endpoints. These and other studies are presented in more detail in section ‘Nutritional epigenomics: how to make sense of what we measure'.
These and other observational studies can be misinterpreted due to confounders; however, they can help to establish associations, which can then be tested by intervention to demonstrate the causality. The observational evidence has been the starting point for the scope of review since it provides a strong suggestion of a measurable effect apparently linked to an earlier exposure. However, while numerous studies have been carried out using animal models – particularly in relation to obesity – for obvious reasons, human clinical interventions are comparatively rare.
Another crucial element in considering long-term results from early exposure is the timeframe/timing and duration of exposure. In many cases, the potential long-term effect of a single intervention is greatest in the developing system (infant or fetus), and the size of the effective dose may be relatively low. However, the ability to predict the precise effect of the intervention in terms of long-term outcome is also low. In adults, the opposite is often the case. The dietary intervention may have to be significant and the prediction of effect is immediately measurable but the long-term effect of a single intervention is low as demonstrated conceptually in Fig. 1.
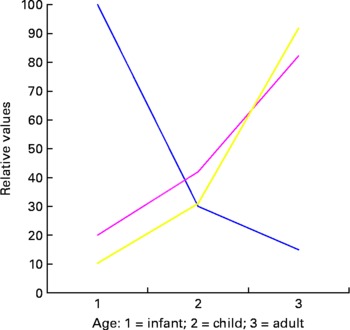
Fig. 1 Conceptual figure on the effects of exposure at different developmental ages. ![]() , predictive power;
, predictive power; ![]() , intervention;
, intervention; ![]() , long-term effect.
, long-term effect.
A further complication is that clinical endpoints change with time and often the effects of the initial programming event may be significantly diluted by a lifetime exposure to a range of other factors. This, together with reducing metabolic plasticity and increased differentiation, may be a major factor in the lowering of programming potential as cells and organisms get older (Fig. 2).

Fig. 2 The shrinking of unmodified endpoints and increase in environmentally modified endpoints over time as external factors impact through a lifetime.
In mechanistic terms, it has been hypothesised that fetal developmental plastic responses can cause changes in lean body mass, endocrinology, blood flow and vascular loading. These responses are then modulated or amplified in infancy and childhood and therefore lead to increased susceptibility– particularly to cardiovascular and metabolic disease – in adulthood(Reference Javaid, Crozier and Harvey9). In developmental terms, fetal exposure to a range of dietary components including Ca, folate, Mg, high or low protein and Zn as a result of maternal diet have all been, with varying degrees of confidence, associated with birth weight. Low birth weight is by itself associated with a range of long-term outcomes, including insulin resistance and type 2 diabetes in later life.
A further addition to the genre was the use of the term ‘metabolic imprinting’ which was adopted by Waterland & Garza(Reference Waterland and Garza10), who suggested that while biological mechanisms exist to ‘memorise’ the metabolic effects of early nutrient exposure, these were not hypothesis driven. They proposed to apply more rigour to the testing of chronic disease outcomes and on the development of biological mechanisms and the testing of hypotheses. Metabolic imprinting was defined as ‘the basic biological phenomena that putatively underlie the relations among nutritional experiences of early life and later diseases’.
The authors also considered that metabolic programming, while being a useful term, did not convey with sufficient clarity the key aspects of imprinting which, as they saw it, was required to encompass both susceptibility limited to a specific developmental window and a persistent effect lasting through adulthood – although it is not clear if the magnitude of the effect should be consistent through adulthood or if a falling off in potency is acceptable.
They also considered that the outcome should be specific and measurable and that a dose–response or threshold relation between a specific exposure and an outcome should be demonstrable. Waterland & Garza(Reference Waterland and Garza10) also distinguished between other types of imprinting (e.g. hormonal and metabolic). The essence of their argument appears to be that ‘imprinting’ has particular characteristics and the term can be used in conjunction with several prefixes dependent upon the target physiological effect but that in each case there should be a mechanistic underpinning for the use of the term.
Following this suggestion, Lucas(Reference Lucas11) raised some questions as to the use of the term ‘imprinting’. His main argument was that programming can encompass a wide range of biological effects, whereas imprinting had a much narrower range. In addition, he also felt that the use of a term more usually associated with a quite distinct event – i.e. gene imprinting, would inevitably lead to confusion. Since this early exchange, a plethora of papers have appeared which seem to use the terms imprinting and programming almost interchangeably. This is not a particularly helpful situation for communication to both scientific and non-scientific stakeholders and a robust and reliable definition is a prerequisite to developing the area.
Epigenetics, on the other hand, has been quite strictly defined in terms of specific molecular events relating to gene expression and provides a mechanistic underpinning for many imprinting/programming events. The development of clear mechanisms to explain the impact of early life exposure on later clinical endpoints would be of great value in predicting the outcomes of specific dietary interventions.
The scope of this review is to lay the foundation for the prioritisation of factors that determine the relative significance of different early exposures in terms of health outcomes. These outcomes should include both mortality and morbidity or quality of life. In this way, we can enumerate the most significant risk factors associated with the early life events and define the causality, association and effects. In particular, to provide a guide for scientists, regulators and policymakers that will enable them to understand what is presently known, prioritise the research to address the gaps and effectively impact upon public health in an understandable and targeted way. Implicit in this analysis is a realisation of the social conditions that pertain to early life exposure and later health and consequences for funding prioritisation.
Enabling technologies and methodology
Biomarkers – what to measure
Biological systems are constantly in a state of flux both due to internal interactions and due to external exposures. In the context of diet and health, biomarkers are factors that reflect biological status at a given time point. For example, dietary stanols and sterols will reduce cholesterol levels in hypercholesterolaemic individuals. High cholesterol is a risk factor for CVD; therefore, blood cholesterol is a biomarker which reflects the increased risk of a disease outcome and may be affected by a specific dietary intervention. Most biomarkers measure biological response at a specific time point and hence the effect of a given intervention at that time point also. Finding relevant, predictive biomarkers related to programming or imprinting is not straightforward. The biologically relevant event remains significant long after the exposure responsible for it has ceased. Finding biomarkers that are not only predictive of a later effect but which, under the best circumstances, remain measurable once the initial exposure has ceased constitutes a major problem. This will become clear when we consider definitions of biomarkers, their validation and how they can best be used.
Biomarkers have conveniently been divided into sub-categories(Reference Branca, Hanley and Pool-Zobel12). The definitions are designed to allow for a simple categorisation but it also tends to describe the analytical methodology used. In addition, it also introduces the concepts of surrogate biomarkers and predictivity.
Exposure
A biomarker of exposure can be defined as a chemical entity (or something derived directly from it), which is measurable in an exposed individual and reflecting that exposure in a dose-dependent fashion. The simplest exposure biomarkers are the components of interest themselves. It is implicit in this type of measurement that the component is either unchanged by metabolism or can be converted into something that is directly measurable.
Since programming implies some alteration in status, exposure biomarkers are only relevant to programming if they are associated with a known and measurable effect in a time period after exposure. The measurement taken early in life may provide useful information concerning the likelihood of a subsequent health endpoint once the exposure has ceased. Observational studies such as ALSPAC in which a specific exposure is correlated with later outcomes are valuable since they can lead to the development of hypothesis-driven research. Many such studies reveal correlations rather than causal associations and this is an inherent weakness of this type of investigation. The utility of biomarkers of exposure in the context of programming is where they are linked to mechanistic or associative knowledge of the consequences of that exposure. The great advantage of developing robust biomarkers of exposure is that they are measurable events at a time when it is possible to change the outcome by dietary manipulation. A typical exposure biomarker could include the levels of dioxins whose presence in the body is reflective of earlier exposure.
Susceptibility
A biomarker of susceptibility has traditionally largely encompassed genetic polymorphisms or variability that give rise to an increased susceptibility to an effect. This can be a direct (genetic) or an indirect effect. The implication is that a biomarker of genetic variability is a distinct and measurable entity in a gene which can be used to predict likely outcomes. Such genes are referred to as ‘imprinted’ genes.
More recently, susceptibility has grown to include epigenetic effects where there is a connection between certain genes, exposure to some environmental (including dietary) factors and later biological events. For example, folate deficiency affects epigenetic events and has been implicated in colon cancer susceptibility. This led to the conclusion that ‘the portfolio of evidence from animal, human and in vitro studies suggests that the effects of folate deficiency and supplementation on DNA methylation are gene and site specific, and appear to depend on cell type, target organ, stage of transformation and the degree and duration of folate depletion’(Reference Kim13). In these terms, susceptibility is not fixed but is measurable at a specific time. It therefore becomes a fluid and dynamic process which is affected by external factors and may change depending on when in the temporal sequence it is measured.
Effect
Biomarkers of effect comprise the most challenging group. In order for a biomarker of effect to be useful in the context of programming, it must fulfil some key criteria.
(1) It should be measurable at a time point when it is able to be altered by an external (dietary) component and that alteration should be reflective of an eventual changed health endpoint. Biomarkers of effect in general may be measured at a time that is distant from the exposure and can also be as a result of cumulative exposure (e.g. DNA damage). However, in order to be relevant to programming, an effect biomarker must be predictive of a future outcome. It should be measurable before and after the exposure event and should predict the biological outcomes.
(2) There must be a dose-related, mechanistic rationale between exposure (and a biomarker of exposure) and the biomarker of effect and to the health endpoint. This means that many biomarkers of effect are measuring surrogate endpoints.
(3) In common, with all biomarkers, it must be validated both in terms of analytical methodology and biological integrity.
Many of the biomarkers of effect and exposure and their relationship to eventual health outcomes have been indicated in the first instance by prospective epidemiological studies. These have suggested associations between certain events and later onset of disease. While these are certainly useful in suggesting new potential areas for study, it is important to recognise that such studies have a high tendency to being blurred by confounders, and the search for a mechanistic underpinning may be futile.
Specificity of human/animal models
Research into developmental programming of adult health and disease has made considerable progress over the past two decades but a clear consensus on the exact nutrients involved and their mechanisms remains to be established. This problem relates in part to the long-term developmental time frame in which changes in metabolism and cardiovascular function occur. At the same time, the discipline has had to contend with the dual challenges of integrating findings from lifelong epidemiological studies which have largely focussed on birth weight and its relationship (or otherwise) with adult disease and our ability to incorporate these results into appropriate nutritional intervention studies using animal models. It is now established that there are many potential influences on offspring outcome. For example, maternal body composition, age and parity, genetic constitution, macro and micronutrient intake and handling, size, shape and number of offspring, sex, type of lactation and so on.
In this section, we attempt to provide an overview of some of the major problems with both human and animal studies in conjunction with optimal experimental paradigms that may be utilised in future research aimed at elucidating the precise mechanisms by which changes in the maternal diet during reproduction can impact on the life time health of resulting offspring. Particular emphasis will be given to the applicability of the animal model that has been utilised and how similarities in the reproductive process may enable its best use in future examinations of the nutritional programming of adult health and disease.
Human models – historical and contemporary models of developmental programming
The majority of the early work conducted by David Barker and colleagues utilised data from historical cohorts born in the 1930 s and benefited from the meticulous birth records, often kept by the same individual over long periods of time(Reference Kim13). This enabled clear relationships between the size, shape and placental mass of an infant at birth with hypertension later in life(Reference Barker, Bull and Osmond14). Subsequently, the recruitment of long-term historical records from Finland has enabled longitudinal studies on infant growth to be related to adult insulin sensitivity(Reference Barker, Osmond and Forsen15). More recently, the use of nutritional interventions of preterm formula in randomised controlled studies has emphasised the impact of inappropriate growth in early infancy on later disease risk(Reference Singhal16). What is apparent, however, from more contemporary studies and the rise in both childhood and adult obesity is the complexity of this process and how changes in both activity and dietary intake make the translation of findings from such studies into present lifestyle interventions very difficult. At the same time, the causes of the ongoing epidemic of obesity and the predicted increase in associated renal and CVD are multifactorial(Reference Keith, Redden and Katzmarzyk17) and these may be either exacerbated or reduced by early dietary exposure(Reference Williams, Kurlak and Perkins18).
Critical developmental stage
One consistent theme that is apparent from both historical and contemporary studies is that changes in nutrition at specific stages of pregnancy can have very different outcomes(Reference Symonds, Stephenson and Gardner19). This is not unexpected as different organs have critical and precise developmental stages which may be compromised, or enhanced, and, thereafter, be permanently set for the rest of that individual's life. Importantly, adaptations of this type appear to be dependent not only on the period in which the mother's diet is altered but also on the diet to which she is rehabilitated(Reference Reynolds, Godfrey and Barker20). One fundamental consideration is the self-limitation in food intake between early and mid gestation that occurs commonly as a result of nausea affecting approximately 90 % of women in the UK that may be directly linked to Western diets(Reference Pepper and Roberts21). The extent to which this directly relates to changes in placental function and/or fetal growth remains less clear but there is a need to match global prenatal and postnatal nutritional requirements so as to avoid accelerated growth (during pregnancy and early infant life)(Reference Gardner, Tingey and van Bon22) and the concomitant increased risk of later obesity and metabolic complications(Reference Symonds23). Importantly, however, intergenerational acceleration mechanisms do not appear to make an important contribution to levels of childhood BMI within the population(Reference Davey Smith, Steer and Leary24).
The lactational environment and postnatal development
A further area requiring consideration is the relationship between the maternal diet in late pregnancy, its impact on mammary gland development and milk production and whether the infant is breast-fed or formula fed(Reference Toschke, Martin and von Kries25). A higher macronutrient content of formula feed compared with breast milk, in conjunction with its fixed composition throughout a feed – unlike in breast-fed infants for whom milk composition changes with time – will impact on nutrient supply to the infant. It is, therefore, not only the short-term but also the long-term advantages of breast-feeding in terms of development of appetite regulation that should be considered in this regard(Reference Sievers, Oldigs and Santer26). The type of lactation also impacts on other behavioural aspects, including sleep–wake activity cycles(Reference Lee27), so that extended breast-feeding may not only be beneficial in developing countries but also in developed countries(Reference Heird28). Other confounding factors such as social class and smoking during pregnancy and lactation further determine postnatal diet(Reference Bogen, Hanusa and Whitaker29).
In summary, the relationship between nutrient supply and the key stages of development from the time of conception to weaning is highly complex and requires careful, in-depth consideration. It is necessary to conduct detailed animal experiments in a range of species in order to elucidate the mechanisms involved, be they epigenetic or related processes(Reference Symonds, Stephenson and Gardner30).
Classic animal models of nutritional programming
The main animal models that have been utilised to date to investigate the impact of maternal diet on long-term programming have been rats and sheep(Reference McMillen and Robinson31). These obviously have very different developmental patterns in not only the relationship between placental and fetal growth but also in maturity at birth and milk composition(Reference Prentice and Prentice32). The advantage of using rats is their very short gestational length. However, the type of diet they consume in the wild is very different to that fed to housed laboratory animals in which semi-purified diets are the norm. Such diets provide substantially greater nutrients to pregnant rats than to controls, and thus should be considered ‘pharmalogical’ as opposed to ‘physiological’ and are well outside the normal distribution. For example, in the case of high-fat diets, these contain four times as much fat compared with control diets and are at risk of being deficient in micronutrients. Not surprisingly, when fed a diet so rich in fat, maternal food intake is reduced(Reference Taylor, McConnell and Khan33). In addition, rats exhibit coprophagia which has a substantial effect on nutrient flux and the ability to experimentally manipulate the intake of specific nutrients. It should be noted, however, that recent rodent models have been developed to overcome the issue of high fat content at the expense of other essential nutrients.
Appreciable placental growth continues up to term in the rat which is necessary in part to meet the much higher protein demands for fetal growth compared with that seen in human subjects or sheep(Reference Widdowson34). In large mammals, the maximal period of placental growth is early in pregnancy and is normally necessary to meet the increased fetal nutrient requirements in late gestation when fetal growth is exponential(Reference Symonds and Gardner35). Furthermore, rats produce large litters whereas sheep and human subjects normally produce only one (or two) offspring of comparable birth weight per pregnancy.
Methodological considerations and the interpretation of metabolic programming in rat and sheep models
There have been two major problems with rat studies with regard to assessment of the long-term cardiovascular outcomes. First, in many studies, blood pressure has only been measured using the tail-cuff technique in restrained and heated animals during the day when they are normally inactive(Reference Kwong, Wild and Roberts36). The results with this method differ considerably from those obtained from telemetry(Reference D'Angelo, Elmarakby and Pollock37). The tail-cuff method was originally validated and recommended to use only in hypertensive animals(Reference Bunag38). Modest differences in blood pressure recorded in normotensive rats are not always informative, which may explain why, in more recent studies, offspring born to dams fed a low-protein/high-carbohydrate diet through pregnancy show either no difference or a reduction in blood pressure when measured using either a telemetry or an indwelling arterial catheter(Reference Fernandez-Twinn, Ekizoglou and Wayman39, Reference Hoppe, Evans and Moritz40). Comparable findings are seen in offspring born to dams in which food intake is reduced by 50 % through pregnancy compared with controls(Reference Brennan, Olson and Symonds41). There also appears to be a marked divergence in the long-term outcomes between sexes in rats that is primarily linked to the faster, as well as continued, growth of males compared with females(Reference Symonds and Gardner35).
Despite the fact that sheep are ruminants, they have proved valuable in enabling us to understand the nutritional and endocrine regulation of placento-fetal development. Like in human subjects, the primary metabolic substrate for fetal metabolism is glucose, for which GLUT 1 is the main placental regulator(Reference Dandrea, Wilson and Gopalakrishnan42). Glucose is, thus, transported across the placenta by active diffusion determined by its concentration in maternal blood(Reference Edwards, Symonds and Warnes43). In addition, not only does kidney development show a very similar ontogeny between sheep and human subjects, but the distribution of total nephrons across the adult population is also comparable(Reference Symonds, Budge, Mostyn and Watkins44). It is also feasible to obtain very consistent blood pressure recordings in the offspring using arterial cannulation while the animal is standing freely with continual access to its diet(Reference Gardner, Pearce and Dandrea45). At the same time, there is no discernable difference in blood pressure control or glucose regulation between sexes when measured in intact adult sheep(Reference Gardner, Tingey and van Bon22, Reference Symonds, Budge, Mostyn and Watkins44).
In summary, the use of both rats and sheep as models for examining the long-term effects of early nutritional interventions is valid and can produce consistent results. Extrapolation of these to the human situation must be carried out with care and with a clear understanding of the discrepancies of both model systems.
Other models
Preterm infants
Preterm-born babies may represent a human model of the third trimester of pregnancy in which the impact of the environment including nutrition can be studied. Although the precise nutritional needs of the fetus to support optimal growth velocity are not known, for instance, amino acid and long-chain PUFA (LCPUFA) supplementation have been shown to improve the early weight gain and/or body composition, respectively(Reference Valentine, Fernandez and Rogers46–Reference Innis, Adamkin and Hall48). However, exposure to other environmental factors associated with being born preterm including high risk of infection and other non-nutritional factors does complicate this further.
Thus, suitable nutritional interventions may now be available that can examine the relevant short- and long-term outcomes in a consistent and validated manner to determine how contemporary diets impact on fat deposition, metabolic homoeostasis and cardiovascular control in animal models. The completion of such studies may enable us to determine the optimum nutrition in terms of quantity and quality. However, as Table 2 demonstrates nutritional interventions, even to demonstrate short-term effects, must control for potential confounders and demonstrate the importance of sound intervention design in this growing field.
Table 2 Summary of studies into nutritional programming in which outcome measures are potentially confounded by a mismatch between groups in their composition of offspring from singleton and twin pregnancies

Nutritional epigenomics: how to make sense of what we measure
Slowing down or preventing the alarming progression of obesity worldwide represents a major public health challenge and a major health concern for future generations. The presence of a heritable or familial component of susceptibility to obesity is well established(Reference Rankinen and Bouchard49). However, apart from extremely rare cases of monogenic forms, most cases correspond to a multifactorial disorder(Reference Rankinen, Zuberi and Chagnon50). This said, however, obesity is a good example of epigenetics as these common forms are associated with a range of genetic and non-genetic familial factors, triggered by the ‘developmental origins of disease’ phenomenon and aggravated by environmental factors, as shown by the rate of discordance between monozygotic twins(Reference Bouchard, Tremblay and Despres51, Reference Fraga, Ballestar and Paz52). Epigenetic misprogramming during development is now widely thought to have a persistent effect on the health of the offspring and may even be transmitted to the next generation(Reference Gallou-Kabani and Junien53).
The term ‘epigenetics’ has been defined as ‘the causal interactions between genes and their products which bring phenotype into being’(Reference Waddington54). It is now used to refer to stably maintained mitotically (and potentially meiotically) heritable patterns of gene expression occurring without changes in DNA sequence. Mechanistically, this is achieved by a range of modifications, including DNA methylation and a complex repertoire of histone modifications: acetylation, methylation, phosphorylation, ADP ribosylation, ubiquitination leading to chromatin remodelling. These processes add to the information of the underlying genetic code conferring unique transcriptional instructions. Epigenetics instructions and machinery create a dynamic nuclear environment that specifies transcriptional states and comprises the essential components of heritable cellular memory, a hallmark of differentiation. Despite sequencing of the human genome studies of the finely tuned chromatin epigenetic networks, DNA methylation and histone modifications are required to determine how the same DNA sequence generates different cells, lineages, organs and ultimately the phenotype.
The mechanism of epigenetic manipulation
DNA methylation patterns and histone modifications are responsive to the environment throughout the life. The epigenetic landscapes are affected by environmental and genetic influences such as embryo culture conditions, DNA methyltransferase 1 overexpression, hyperhomocysteinaemia and folate deficiency either before or during pregnancy, in the postnatal and post-weaning period that persist into adulthood. Transient nutritional stimuli occurring at critical ontogenic stages may have long-lasting influences on expression of various genes by interacting with epigenetic mechanisms and altering the chromatin conformation and transcription factor accessibility.
Several types of sequences associated with specific epigenetic makeup are targets of a host of environmental factors that can trigger transiently or permanently disturbed chromatin architecture with altered epigenetic instructions – either at the somatic or at the germline levels – leading to aberrant patterns of gene expression.
For example,
(1) unique genes, e.g. the glucocorticoid receptor, or, more likely, specific subsets of unique genes belonging to different pathways or systems(Reference Weaver, Cervoni and Champagne55, Reference Champagne, Weaver and Diorio56);
(2) genes present as multiple copies such as of genes coding ribosomal RNA(Reference Santoro57, Reference Oakes, Smiraglia and Plass58);
(3) whole genome epigenomic changes(Reference Pham, MacLennan and Chiu59–Reference Pogribny, Ross and Wise61).
However, still little is known about the various replication/DNA synthesis-dependent and -independent epigenetic mechanisms underlying the stochastically, genetically and environmentally triggered epigenetic changes occurring during an individual's lifetime. They may result from replication-dependent, replication-independent or DNA repair events. Most of the epigenetic changes were thought to be coupled to DNA replication. Thus, epigenetic patterns need to be faithfully maintained during each cell cycle. In addition, the maintenance of genome integrity involves specific repair pathways(Reference Polo, Roche and Almouzni62). During the synthesis phase, this is achieved by duplication of chromatin structure in tight coordination with DNA replication. Histone synthesis and deposition onto DNA by chromatin assembly factors ensures efficient coupling with DNA synthesis(Reference Nakatani, Ray-Gallet and Quivy63). However, this faithful maintenance is not always required. Changes in epigenetic patterns are observed during the differentiation processes for several genes involved in development, cellular growth, differentiation, apoptosis or tissue- or sex-specific expression. DNA demethylation modulates mouse leptin promoter activity and the insulin-sensitive GLUT4(Reference Yokomori, Tawata and Onaya64) during the differentiation of 3T3-L1 cells (mouse embryonic fibroblast – adipose-like cell line)(Reference Yokomori, Tawata and Onaya64, Reference Yokomori, Tawata and Onaya65).
The evidence for epigenetic systems
Recently, links have been found between circadian rhythms and major components of energy homoeostasis, thermogenesis and hunger–satiety, rest–activity rhythms and the sleep–wake cycle(Reference Staels66, Reference Fontaine and Staels67). The rhythmic, circadian induction of a substantial proportion of genes, by a network of clock genes, one of which is a histone acetyltransferase, by nuclear receptors and transcription factors is also controlled by chromatin remodelling. The associated circadian epigenetic patterns must be replication-independent, transient, sensitive to environmental cues and reversible. However, poorly adapted behaviour or lifestyle and desynchronised cues may disturb the modulation of gene expression. This may ultimately lead to persistence of aberrant and unphased ‘locking’ or ‘leakage’ of gene expression and unadapted responses of the organism in terms of physiology, metabolism and behaviour to environmental changes. Thus, epimutations accumulate over time, increasing the ‘epigenetic burden’ potentially leading to the onset of age- and/or environment-related diseases(Reference Issa68). The lifelong remodelling of our epigenomes by nutritional metabolic and behavioural factors corresponds to the new field of ‘nutritional epigenomics’.
Trans-generational effects
It is now widely accepted that the developmental basis of adult diseases and the non-Mendelian transmission of acquired traits cannot be attributed solely to genetic mutations or a single aetiology(Reference Whitelaw and Whitelaw69). In addition, there is accumulating evidence that during the periconceptual, fetal and infant phases of life, exposure to environmental compounds or behaviours, placental insufficiency, maternal inadequate nutrition and metabolic disturbances can promote improper ‘epigenetic programming’, leading to susceptibility to various disease states or lesions in the first generation and sometimes subsequent generations, i.e. transgenerational effects. While developmental programming may imply an altered uterine milieu perpetuating the disease risk through the cycle of mother-to-daughter transmission, with epigenomic alterations at the somatic level, there are also examples of transmission through the germline for both sexes and with sexually dimorphic effects(Reference Gluckman, Hanson and Beedle70, Reference Yang, Schadt and Wang71). There are an increasing number of animal models, designed to mimic human conditions, that clearly involve an epigenetic and/or gene expression-based mechanism and these have recently been reviewed(Reference Junien and Nathanielsz72).
DNA methylation alterations and/or histone modifications involve different types of sequences either at the somatic or at the germline levels. However, very little information is presently available to evaluate the actual impact, persistence, and dietary and therapeutic reversibility of these environmentally triggered transgenerational effects. It remains difficult to determine the conditions required for the persistence of transgenerational effects over several generations, even in the absence of the original stimulus. It also remains unclear whether the continuation of exposure over several generations leads to ‘locked’ epigenomic patterns. If this were the case, permanently methylated cytosines, with their higher rate of mutation, would give rise to genuine genetic mutations, thereby persisting in the genome. This would have important consequences for adaptation to new environments – coping with the worldwide epidemic of obesity, for instance.
Epigenetic studies in human subjects
Recent studies also suggest that part of the epigenetic component can be dependent on genetic changes: there is a genetic basis for epigenetic variability between individuals, in stochastic events, susceptibility to environment/diets, to replication-dependent and replication-independent events. The finding that DNA methylation profiles can be associated with particular alleles is of considerable interest. Only a few studies in human subjects have identified associations between DNA sequence and epigenetic profiles. The present population-based approach to common diseases relates common DNA sequence variants to either disease status or incremental quantitative traits contributing to disease. This purely genetic approach is powerful and general, and Bjornsson et al. (Reference Bjornsson, Danielle Fallin and Feinberg73) have proposed an approach to incorporate epigenetic variation into genetic studies. Indeed, it could be that epigenetic variation (including at the epiallele and epihaplotype) may be a better predictor for risk of disease, including late onset and progressive nature of complex diseases than sequence-based approaches alone(Reference Abdolmaleky, Smith and Faraone74, Reference Petronis75).
Future studies in epigenetics
Depending on the nature and intensity of the insult, the critical spatiotemporal windows and developmental or lifelong processes involved, these epigenetic alterations can lead to permanent changes in tissue and organ structure and function; alternatively, some of the gene- and/or tissue-specific changes can be reversible by means of appropriate epigenetic tools. Given several encouraging trials, prevention and therapy of age- and lifestyle-related diseases by individualised tailoring to optimal epigenetic diets or drugs are conceivable(Reference Egger, Liang and Aparicio76, Reference Ou, Torrisani and Unterberger77). However, these potential interventions will require intense efforts to unravel the complexity of these epigenetic, genetic, stochastic and environmental interactions and to evaluate their potential reversibility with minimal side effects. Given the significant and increasing proportion of women who are overweight and overfed when pregnant paying attention to the over-nourished fetus is as important as investigating the growth retarded one(Reference Muhlhausler, Adam and Findlay78). Improving the environment to which an individual is exposed during development may be as important as any other public health effort to enhance population health worldwide(Reference Gluckman, Hanson and Beedle70). It is clear that epigenetic alterations can no longer be ignored in evaluations of the causes of obesity and its associated disorders. There is a need for systematic large-scale epigenetic studies on obesity, employing appropriate strategies and techniques and appropriately chosen environmental factors during critical spatiotemporal windows in development.
Perinatal nutrition and CVD in adults
Background to diet effects
The possible impact of perinatal nutrition (including both in utero nutrition and lactation) on the development of CVD in adulthood is a very complex issue, mainly because of the very slow evolution of the disease before clinical manifestation. The progression of fatty streaks to coronary atherosclerosis and then to the stenosis that will provoke cardiac ischaemia or infarction may require 50 years. Moreover, the possible evolution of infarct (or hypertension) to cardiac hypertrophy and/or chronic heart failure may also require a long duration. This cardiac disease ‘continuum’ has been related to several risk factors, including cholesterol levels, diabetes, obesity, sedentarity, coagulation, smoking, dietary practices and vascular dysfunction. In this context, it is difficult to evaluate the possible influence of the perinatal environment, including maternal factors (genotype, nutrition, disease state including dyslipidaemia, gestational diabetes and hypertension), fetal predisposition (genotype development) and lactation. It is obviously difficult to differentiate these early putative risk factors from those which develop later independently of the link from childhood to adulthood.
Epidemiological evidence of association
Hypertension
Birth triggers the transition from a low blood pressure system to a high blood pressure system. Intra-uterine undernutrition is known to affect later hypertension both in experimental animals and in human subjects and the mechanism has been investigated(Reference Franco, Akamine and Di Marco79). Intra-uterine undernutrition impairs nephrogenesis and glomerular hypertrophy. These developmental alterations induce decreased filtration rate and a decreased plasma flow that will contribute to increased blood pressure. Besides this kidney functional alteration, intra-uterine undernutrition also affects the endothelium function. The mechanism involves a reduction of superoxide dismutase activity, an increase in NADPH oxidase activity and a decrease nitric oxide synthase gene expression and activity. As a consequence, nitric oxide production is reduced, whereas free radical oxygen is increased, resulting in a change in relaxation of vascular smooth muscle cells.
The quality of lactation was also shown to affect blood pressure in human subjects. Diastolic and mean blood pressure at the age 13–16 years were significantly lower in children previously fed banked breast milk compared with children fed either term or preterm infant formulas. Moreover, the authors report than the results remained unchanged after adjustment for present BMI, sex and Na intake(Reference Singhal, Cole and Lucas80). However, this result was not confirmed in large-scale epidemiological studies. The Oxford Nutrition Survey investigated pregnant women recruited in 1942–4 to determine whether the wartime dietary rations were sufficient to prevent the deficiencies. More than 50 years later, the offspring were recruited to explore the possible impact of maternal nutrition in pregnancy on CHD risk factors, including blood pressure, but the results provided no evidence to support the hypothesis that birth weight or undernutrition in pregnancy affect hypertension(Reference Huxley and Neil81). Similarly, The Boyd Orr Cohort investigated a cohort of children born 1937–9 and their follow-up in 732 adults aged 65 years and this reported no evidence of the influence of breast-feeding on blood pressure(Reference Martin, Ebrahim and Griffin82).
Athersclerosis
The Boyd Orr Cohort, comprising 700 adults between in the years 1937 and 1939 (see above), was recently reinvestigated. The authors report that the breast-fed group displayed lower intima-media thickness of carotid arteries and a lower score in carotid and femoral plaques(Reference Martin, Ebrahim and Griffin82). The results remained unchanged after adjustment for socio-economic variables (including smoking and alcohol), and adjustment for pathway causal factors (including blood pressure, adiposity, cholesterol, insulin resistance and C-reactive protein). Atherosclerosis is considered to begin very early in life as shown in the Fate of Early Lesions in Children Study(Reference Napoli, Glass and Witztum83). This study showed that maternal hypercholesterolaemia during pregnancy induces changes in fetal aorta that may determine the long-term susceptibility of children to fatty-streak formation and subsequent atherosclerosis. The human fetus displays arterial fatty streaks in utero. Although these fatty streaks regress after birth, they redevelop rapidly independently of the cholesterol status of the child. The study reports that these fatty streaks are associated with an increase in arterial wall thickness (aorta and carotids) in child than in fetus. Investigations in animals(Reference Palinski, D'Armiento and Witztum84) showed similar results, the offspring of hypercholesterolaemic mothers displaying a significantly higher atherosclerosis lesion score at birth, at 6 months and at 12 months. Interestingly, when the mothers were treated with cholestyramine during pregnancy, the atherosclerosis lesion score in the offspring was significantly lower at birth and at 6 months and fully normalised at 12 months(Reference Huxley and Neil81). However, although several nutrients, including phytosterols, the SFA:PUFA ratio and n-3 PUFA, affect cholesterol transport in adults, the impact of these nutrients in early development has not been considered so far.
Myocardium and coronaropathies
The Helsinki Birth Cohort Study including more than 4000 men born 1934–4 reported a significant correlation between the ponderal index at birth (term babies only), early growth and the standardised mortality ratios for CHD(Reference Eriksson, Forsen and Tuomilehto85). Low birth weight and low ponderal index were associated with increased CHD. After 1 year of age, rapid gain in weight and BMI increased the risk of CHD in those men with a low ponderal index at birth. Epidemiological studies can be confusing. Investigations in the ‘Nurses' Health Study’ cohort suggested that breast-feeding may be associated with a reduction in risk of ischaemic CVD in adulthood(Reference Rich-Edwards, Stampfer and Manson86). Conversely, investigations on the ‘Caerphilly study’ cohort data provide little evidence of a protective influence of breast-feeding on CVD risk factors, incidence or mortality. Moreover, a possible adverse effect of breast-feeding on CHD incidence was reported, which may be related to the difficulties in differentiating the lactation effects from the individual risk factors developed after weaning(Reference Martin, Ben-Shlomo and Gunnell87).
In the perinatal period, the myocardium is subjected to several key changes and some of these changes will induce a phenotype influencing cardiac function. These could be the key parameters in the pathology developed in later life such as mitochondria oxidative capacity and adrenergic regulation of cardiac function, both through membrane phospholipid homoeostasis.
Critical developmental stage
The question is then restricted to the developmental stage during which the specific impact of the perinatal nutrition period on the development of CVD, independently from the known risk factors, developed during independent life. This may affect hypertension, atherosclerosis development and localisation, individual sensitivity to ischaemia and preconditioning, occurrence and severity of infarct, development of cardiac hypertrophy and its evolution in chronic heart failure.
Several organ systems which can subsequently influence cardiovascular function via programming mechanisms have been documented in human subjects. These include the vessels (vascular compliance and endothelial function), the endocrine system (glucose and insulin metabolism), the muscles (glycolysis in exercise and insulin resistance), the kidneys (rennin–angiotensin system) and the liver (cholesterol metabolism, fibrinogen and factor VII)(Reference Godfrey and Barker88). However, all these investigations referred specifically to perinatal dietary restriction and raise the issue of qualitative concern. Moreover, the data on the heart itself are scarce and the role of metabolic programming that may affect cardiac metabolism and function is unclear.
Mitochondria oxidising capacity
The transition of the cardiomyocyte energy production system from exclusive glucose oxidation to fatty acid (FA) oxidation allows the large increase in cardiac energy production capacity as required by independent life. This process is based on a large increase in mitochondria mass controlled by several key factors including mitochondrial DNA (always from maternal origin) mainly encoding for the electron transport chain, transcriptional co-activator PGC-1α(89) which controls mitochondrial biogenesis, the development of FA oxidation pathways(Reference Huss and Kelly90, Reference Huss and Kelly91)and also cardiolipin (CL) synthesis through PPAR. Feeding dam rats a high-fat diet during pregnancy resulted in offspring which at 6 months display a significant decrease in mitochondria encoding mRNAs (mainly cytochrome oxidase subunits, dicarboxylate carrier and mitochondrial genome)(Reference Taylor, McConnell and Khan33). CL is the key phospholipid in the function of inner mitochondrial membrane ensuring the cohesion of the electron transport chain and associated enzymes. At the cellular level, cardiac ischaemia is basically a crisis of energy production associated with an unbalanced ratio in substrate oxidation (excessive FA oxidation and decreased glucose oxidation)(Reference Grynberg92). This unbalanced metabolism contributes to the rapid oxidation of CL which decreases in mitochondrial membranes, impairing energy production(Reference Paradies, Petrosillo and Pistolese93). The cardiac capacity to restore CL is partly controlled by the effect of LCPUFA on PPAR, and maternal diet was reported to influence the acyl composition of CL (and hence its sensitivity to oxidation) via both FA placental transfer and breast milk(Reference Berger, Gershwin and German94). However, chronic heart failure is associated to a decreased capacity of the cardiomyocytes to produce energy from FA resulting in a reduced capacity to face any increase in energy demand (the term ‘metabolic regression to fetal phenotype’ is often encountered in the literature)(Reference Huss and Kelly91). The efficiency of mitochondrial biogenesis and CL synthesis and the basal mitochondrial mass are key factors in cardiac pathophysiology.
Cardiac function
The perinatal period is also associated with the transition in the neurohumoral regulation of cardiac function to a large predominance of the β-adrenergic system. This system involves the internalisation of the receptor and its recycling to sarcolemma (clathrin-mediated recycling) in which phosphatidylinositol-3-kinase plays a key role. The use of β-blockers in the treatment of cardiac disease including coronaropathy and chronic heart failure outlines the importance of the basal β-adrenergic function. The development of this pathway is based on the membrane homoeostasis of phosphatidylinositol, the substrate of phosphatidylinositol-3-kinase. This enzyme is also involved in insulin signalling by triggering the translocation of GLUT4 to the membrane. The early development of the phosphatidylinositol-3-kinase pathway may thus impact on both neurohumoral regulation of cardiac function and insulin control of cardiac metabolism, since insulin contributes to myocardium substrate balance through the regulation of AMP-activated kinase-like leptin and adiponectin.
Experimental evidence and mechanistic understanding
Several attempts have been made, using animal models, to investigate the cardiac consequences of in utero nutrition. The effect of maternal undernutrition during pregnancy was studied using sheep as a model system. Dong et al. (Reference Dong, Ford and Fang95) reported a change in the expression of insulin-like growth factor (IGF-1), IGF-1R and IGF-2R, in fetal myocardium associated with a ventricular enlargement in fetus. Han et al. (Reference Han, Austin and Nathanielsz96) reported several alterations of gene expression and particularly the upregulation for several proteins that have been linked to cardiac hypertrophy and compensatory growth in several species including human subjects. Other authors reported in the same model that maternal undernutrition decreased immunoreactive type 1 and type 2 angiotensin-II receptors (AT1 and AT2) in the left ventricle of the fetuses without affecting gene transcription of the angiotensin-II receptors or increased the transcription of mRNA for vascular endothelial growth factor, whereas immunoreactive vascular endothelial growth factor remained unchanged. All together, these data suggest a relationship between maternal undernutrition and later cardiac remodelling processes. The rat is another frequently used model. Studies have included the link between maternal dietary isoflavones and the sensitivity to dilatation and chronic heart failure of offspring, the influence of litter size on cardiac neurohumoral control, the influence of maternal nutrition on cardiomyocyte length which affects left ventricle capacity in overload and the effect of high-fat diet in mothers on mitochondrial DNA expression.
The role of specific nutrients possibly involved in metabolic imprinting of blood pressure and dietary fat has been investigated. In animal experiments (rats), Khan et al. (Reference Khan, Taylor and Dekou97) reported an increase in systolic and diastolic blood pressure in the adult offspring of dam fed a high-SFA diet during pregnancy. Interestingly, this increase was observed in female offspring but not in males. Investigations on the mechanism showed a significant alteration of endothelial function associated with a modified arterial lipid composition, which could result from a misbalanced SFA:PUFA ratio during early growth(Reference Ghosh, Bitsanis and Ghebremeskel98). Such an effect of LCPUFA was also reported in children. Forsyth et al. compared three groups of infants fed an infant formula or the same formula supplemented with arachidonic acid and DHA or breastfed (also providing arachidonic acid and DHA). After weaning, the children were allowed to return to a non-controlled diet and re-examined 6 years later. The results showed a lower diastolic and mean blood pressure in those children who received LCPUFA during lactation either by breastfeeding or by supplementation.(Reference Forsyth, Willatts and Agostoni99) The mechanism is still unknown but could be related with the differential effect of each LCPUFA on blood pressure according to hypertension aetiology as reported in animal models.
Conclusion and future directions
In conclusion, metabolic programming of the cardiovascular system cannot be considered yet as proven, in spite of several promising results. The range of animal investigations does not suggest a trend in the relationship between perinatal nutrition and adult cardiac function, and/or protection can be considered to be confirmed. Epidemiological studies remain unconvincing and often controversial. In addition, so far, they cover only the domains of maternal food restriction and breast-feeding. In between, there is a strong requirement for mechanistic investigations to provide science-based information on the influence of maternal diet on offspring heart function and protection from later disease. Also, these investigations will have to include the globally misbalanced dietary habits (low protein and high fat) as well as the influence of specific nutrients such as specific FA, glycaemic index, amino acids, salt and minerals, sterols or phytohormones.
Role of perinatal leptin in obesity risk/incidence in adults
Background to diet effects
The incidence of obesity, defined as a BMI >30 kg/m2, is rapidly increasing all over the world(Reference Rodgers, Vaughan and Prentice100). The epidemic now affects young children and accumulative evidence suggest that, in part, the origin of the disease may be influenced by fetal development and early life. Nutritional and hormonal status during pregnancy and early life could interfere irreversibly on the development of the organs involved in the control of food intake and metabolism and particularly the hypothalamic structures responsible for the establishment of the ingestive behaviour and regulation of energy expenditure.
The mechanisms responsible for this developmental programming remain poorly documented. While obesity is a multi-factorial problem and is affected by several factors, recent research indicates that the adipokine leptin plays a critical role in this programming(Reference Cottrell and Ozanne101).
Leptin sources and biological functions
Leptin is produced essentially by the adipose tissue and its plasma levels reflect the fat reserves. There are also several extra adipose sources of leptin. The placenta in human subjects (but not in all species) produces leptin and constitutes an appreciable source of leptin for the fetus during pregnancy(Reference Laivuori, Gallaher and Collura102). The mammary gland is also able to produce leptin, particularly during the early phase of lactation(Reference Smith-Kirwin, O'Connor and De Johnston103). In addition, the mammary gland is involved in the transport of leptin from the mother to the milk, which is an additional source of leptin for the newborn(Reference Uysal, Onal and Aral104). The immature gastrointestinal tract may allow leptin to enter the circulation, but it is likely that leptin may have considerable effects locally and play a role in the maturation of the epithelial lining of the gut. There are leptin receptors present in the gastrointestinal tract, which suggests a localised function. Although initially there may be leptin leakage to the circulation because of gastrointestinal immaturity, access of leptin to the circulation may be limited.
Leptin is involved in an extensive number of biological functions and several isoforms of the receptor have been identified in different organs. The best-known effects are those exerted at the hypothalamic level where the long form of the leptin receptor is predominantly expressed and exerts a pivotal role in the regulation of food intake. In peripheral organs, the short form of the leptin receptor is the dominant form expressed and its biological effects include cell proliferation and cell differentiation in adipose tissue, pancreas, liver, kidney, arteries and immune cells.
Leptin and regulation of food intake
At the hypothalamic level, after crossing the brain–blood barrier, leptin interacts with a complex neuronal network integrating a wide range of nervous, nutritional and hormonal signals. Leptin interacts primarily with the arcuates nucleus, where it inhibits the activity of neurons expressing orexigenic peptides (neuropeptide Y and Agouti-related protein) connected to the lateralhypothalamic nucleus which is recognised as a major controlling factor in hunger. Leptin also stimulates the activity of anorexigenic neurons expressing pro-opiomelanocortin connected to ventromedial nucleus recognised as the centre of satiety. The different hypothalamic nuclei are interconnected via a complex neuronal network with paraventricular and dorsomedial nuclei and the integration of all these stimuli determines the food intake behaviour(Reference Friedman and Halaas105).
Epidemiological evidence for association
There is little epidemiological evidence for an association between circulating leptin and obesity. To briefly summarise, there is a rare leptin gene mutation that causes obesity in early childhood. Small-for-gestational age and preterms have lowered leptin levels. Family history of obesity has been correlated with high umbilical cord levels of leptin.
Critical developmental stage
Leptin stimulation results in a decrease in food intake, and it was initially hoped that exogenous leptin therapy might induce satiety and weight loss in the obese human. Unfortunately, it has been found that obesity is often associated with a ‘leptin resistance’, which is progressively established during ingestion of a hypercaloric diet and associated with an increase of serum leptin levels. The mechanisms underlying leptin resistance remain a matter of debate, but the two hypotheses that have received the most attention are a failure of circulating leptin to reach its target cells in the brain or a blockage of leptin signalling by activation of suppressor of signalling (SOCS3) or specific phosphatases (PTP1b). An alternative hypothesis suggests that leptin resistance may in fact be programmed during fetal and neonatal life and may be the result of an altered development of neuronal circuitry involved in food intake regulation.
The neuronal network is established during hypothalamic development occurring postnatally in rodents. In the mouse, dense neuronal fibre originating from the arcuate nucleus and reaching lateral and dorsomedian hypothalamus and paraventricular nucleus are progressively established between days 6 and 16. The development of this neuronal network occurs at the same period as a dramatic increase in leptin level occurs in the blood, the suggested origin of which is adipose tissue. This change in leptin levels is not related to food intake regulation since body weight of the animals rapidly increases at this period. Bouret, in the group of Simerly(Reference Bouret, Draper and Simerly106, Reference Bouret and Simerly107), clearly demonstrated that leptin at this period exerts a potent neurotrophic action. These authors have observed that ob/ob mice, genetically deficient in leptin, have an altered hypothalamic development characterised by a dramatic decrease in neuronal fibre density in the hypothalamic structures. Secondly, they elegantly demonstrated that leptin administration to these animals during the early postnatal period restored neuronal organisation of hypothalamic circuits in term of fibre density in the paraventricular nucleus. Finally, hypothalamic connections in the diet-induced obese rat model were shown to be permanently disrupted(Reference Bouret, Gorski and Patterson108).
Experimental evidence and mechanistic understanding
Several animal models have clearly shown that either severe undernutrition during pregnancy or placental deficiency, induce intra-uterine growth retardation (IUGR) leading to low birth weight which is associated with low leptin levels. It is also well established that the IUGR newborn show an increased susceptibility to develop obesity and metabolic syndrome when submitted to high-caloric diet during later life. One possible explanation is that leptin deficiency in IUGR causes improper programming. Supporting this hypothesis, it has been recently demonstrated(Reference Vickers, Gluckman and Coveny109) that neonatal leptin treatment of IUGR pups reverses developmental programming induced by mother's severe undernutrition and restores normal adult phenotype. To extend these findings to normal birth weight animals and taking advantage of the recent development of specific leptin antagonist(Reference Solomon, Niv-Spector and Gonen-Berger110), we have recently analysed the consequences of the blockage of the postnatal leptin surge in newborn rats(Reference Attig, Solomon and Taouis111). Leptin mutants (L39A/D40A/F41A/I42) which bind to the leptin receptor with an affinity identical to wild-type leptin, but are completely devoid of agonistic activity, have been administered during early postnatal days 2–13. Three months later, the animals were given a leptin challenge and these animals injected with the leptin antagonist early in life were leptin resistant. When a high-energy diet was given to these animals, they showed a higher susceptibility and a greater increase in body weight than control animals. At 8 months, the animals presented a higher adiposity associated with hyperleptinaemia. These data demonstrate that perinatal leptin in a normal situation plays a crucial role in the determination of the capacity of the animal to respond to leptin later in life and to protect the newborn against the adverse effect of hypercaloric diet.
Several studies have documented the evolution of leptin levels during pregnancy in normal and IUGR babies. It has been shown that leptin levels increased during late fetal life, and at birth, leptin levels are lower in IUGR babies than in normal weight newborn(Reference Jaquet, Leger and Levy-Marchal112). The chronology in the development of the different organs varies between animal species. In contrast to the rodent, the major part of neuronal development occurs before birth in the human(Reference Grayson, Allen and Billes113). However, it is evident that a relative neuronal plasticity remains after birth and this could be even more so in the case of IUGR, where an impairment in the development of several organs (particularly the kidney) is generally observed. As we mentioned earlier, the mammary gland is able to produce leptin and also to transfer leptin from mother's blood to milk. An interesting possibility is to consider that leptin absorbed by the newborn via the milk may be an important factor, which participates in the final maturation of different organs such as the intestine and also hypothalamic structures involved in food intake regulation. Several studies seem to support this hypothesis. Indeed, it has been reported that breast-fed infants have higher serum leptin levels than formula-fed infants(Reference Savino, Nanni and Maccario114) and that breast-fed infants may show an decreased risk of developing obesity(Reference von Kries, Koletzko and Sauerwald115).
Experimentally, it has been demonstrated that the intake of physiological doses of leptin during lactation in rats prevents obesity in later life(Reference Pico, Oliver and Sanchez116). All these facts support the idea that milk leptin may play a favourable role in developmental programming and constitute a credible candidate to explain, at least partially, the protective effect of breast-feeding against obesity. This is an interesting subject for future investigations. In mice, the leptin responsible for the hypothalamic development of the hypothalamic food intake circuitry is thought to be of adipose tissue origin of the newborn despite its very small quantity.
Conclusions and future directions
All the data summarised in this paper suggest that leptin constitutes a key hormonal player during the perinatal period in the prevention of unfavourable developmental programming. Additional basic research is necessary to establish the biological mechanisms involved. In addition to classical rodent models, animal models including sheep or pigs may be useful as models of the human situation. Indeed, as in human subjects, the major part of neuronal hypothalamic development occurs before birth in these two species, although it should be noted that, in the human, major synaptic proliferation occurs after birth. Epigenetic modulations are probably involved in this developmental process and the genes implicated remain to be established.
Different strategies could be envisaged to optimise the effects of leptin during developmental programming. Particular attention must be given to nutrition during pregnancy. Development of well-adapted diets associated with optimised maternal leptin levels would be beneficial.
During the postnatal period, several months of breast-feeding must be encouraged, particularly in IUGR babies. Opportunities for research on the optimisation of the postnatal diet during the critical developmental stage could potentially focus on leptin and its addition to infant formula, for instance.
Perinatal nutrition and type 1 diabetes in adults
Background to diet effects
Type 1 diabetes (T1D), a chronic inflammatory disease caused by a selective destruction of the insulin-producing β-cells of the pancreas, is one of the most common and serious chronic diseases in children(Reference Green and Patterson117, Reference Onkamo, Vaananen and Karvonen118). The incidence is increasing by 3 % per year, particularly in young children and in developed countries(Reference Onkamo, Vaananen and Karvonen118).
T1D is preceded by a pre-clinical phase characterised by autoimmunity against pancreatic islets(Reference Eisenbarth119). A genetic susceptibility for developing islet autoimmunity and T1D is well documented and an environmental influence is assumed(Reference Atkinson and Eisenbarth120).
Epidemiological evidence for association
Over the last 15 years, several groups have initiated prospective studies from birth investigating the development of islet autoimmunity and diabetes(Reference Ziegler, Hillebrand and Rabl121–Reference Honeyman, Coulson and Stone124). These studies provide an opportunity to investigate the factors that are associated with the development of islet autoimmunity and progression to T1D. Findings from these studies have significantly contributed to our present understanding of the pathogenesis of childhood diabetes. However, the exact aetiology and pathogenesis of T1D are still unknown.
Genetic factors influencing the development of islet autoimmunity and type 1 diabetes
Children with a first-degree relative with T1D have a more than tenfold higher risk to develop T1D, further increasing if both parents were affected. Genetic variability in the human leucocyte antigen region explains approximately 50 % of the familiar clustering(Reference Risch125, Reference Davies, Kawaguchi and Bennett126); other genes have also been identified as providing more modest contributions to risk(Reference Davies, Kawaguchi and Bennett126, Reference Cox, Wapelhorst and Morrison127). The concordance of T1D between monozygotic twins is up to 50 %, whereas between dizygotic twins, it is only 10 %(Reference Kyvik, Green and Beck-Nielsen128). Although such differences in the concordance rates between identical and non-identical twins clearly underline the impact of genes on the development of T1D, they also show that genetic susceptibility alone cannot be the ultimate cause for the disease and that environmental factors seem to modify the risk for islet autoimmunity and T1D.
Environmental factors influencing the development of islet autoimmunity
Prospective studies from birth have demonstrated that islet autoimmunity occurs very early in life. Around 4 % of offspring of parents with T1D in the BABYDIAB (genetic risk of developing T1D) study and about 6 % of genetically at-risk infants from the general population in the Finnish Diabetes Prediction and Prevention study have developed islet autoantibodies by age 2(Reference Hummel, Bonifacio and Schmid129, Reference Kimpimaki, Kulmala and Savola130). Children who develop autoantibodies within the first 2 years of life are those who most often develop multiple islet autoantibodies and progress to T1D in childhood(Reference Hummel, Bonifacio and Schmid129). These findings implicate environmental factors that are encountered before age 2 may be important for the development of islet autoimmunity. Candidate environmental factors that are suspected to influence risk for islet autoimmunity in genetically susceptible individuals are dietary factors and factors associated with maternal diabetes.
Critical developmental stage
There are several dietary factors that are proposed to be associated with the development of islet autoimmunity and T1D but most of the research done in this field led to controversial results. There are only few prospective case–control, cohort and human-intervention studies that can be used for hypothesis testing. However, dietary factors that have already been related to the development of islet autoimmunity and T1D were examined in these following prospective studies (Fig. 3). Recently, weight gain in early life was proposed to predict the risk of islet autoimmunity in children with a first-degree relative with T1D(Reference Couper, Beresford and Hirte131).
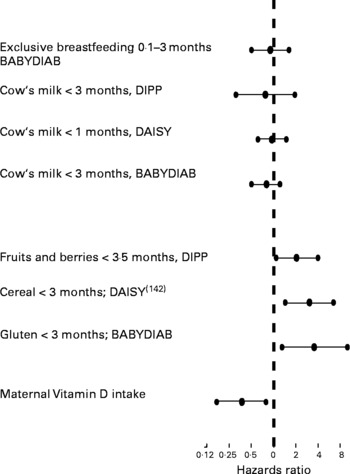
Fig. 3 Studies of the hazards ratios for risk of T1D
It has been suggested by some investigators that breast-feeding may protect against T1D(Reference Sadauskaite-Kuehne, Ludvigsson and Padaiga132), whereas early introduction of supplementary milk feeding may promote the development of islet autoantibodies and T1D(Reference Vaarala, Knip and Paronen133). Four prospective studies in at-risk neonates have not demonstrated an increased risk for developing islet autoantibodies in children who were not breast-fed and received cow's milk (CM) proteins early in life(Reference Couper, Steele and Beresford134–Reference Virtanen, Kenward and Erkkola137). However, recent results suggest that an enhanced humoral immune response to various CM proteins in infancy is seen in a subgroup of those children who later progress to T1D. The authors imply that a dysregulated immune response to oral antigens may be an early event in the pathogenesis of T1D(Reference Lupopajarvi, Savilahti and Virtanen138).
Another candidate factor is the early introduction of solid food in an infant's diet. In two recent prospective studies, it was suggested that risk of development of islet autoimmunity is increased in children who were exposed to cereal proteins, and particularly gluten, early in life(Reference Ziegler, Schmid and Huber139, Reference Norris, Barriga and Klingensmith140). The BABYDIAB study looked at the impact of food supplementation during the first 3 months of life on the development of islet autoimmunity in offspring of parents with T1D. Children who received gluten-containing supplements during the first 3 months of life had a significantly higher risk of developing islet autoimmunity compared with children who received nongluten-containing solid food, CM-based supplements or who were breast-fed only. The Finnish Diabetes Prediction and Prevention study showed that the introduction of fruits and berries before 4 months of life was associated with a significantly higher risk of developing islet autoimmunity compared with children who received solid food supplements later in life(Reference Virtanen, Kenward and Erkkola137).
Dietary factors that have been proposed to protect from islet autoimmunity are vitamin D and n-3 fatty acids. The prospective Dietary Autoimmunity Study in the Young showed that dietary maternal intake of vitamin D was significantly associated with a decreased risk of islet autoimmunity appearance in offspring who were at increased risk for T1D (including a dose–response effect!). Neither vitamin D intake via supplements nor the n-3 and n-6 fatty acids intake via supplements during pregnancy was associated with the appearance of islet autoimmunity in offspring(Reference Fronczak, Barón and Chase141, Reference Zipitis and Akobeng142).
Experimental evidence and mechanistic understanding
There are several ongoing dietary intervention trials in newborns at high risk for T1D:
Trial to reduce type 1 diabetes in the genetically at risk (TRIGR)
To study the impact of CM proteins in an infant's diet on the development of islet autoimmunity and T1D, an interventional trial, the trial to reduce T1D in the genetically at risk, is presently ongoing in children with increased genetic risk and who have a first-degree relative with T1D(Reference Sadeharju, Hamalainen and Knip143). The trial has a double-blind, prospective, placebo-controlled intervention protocol, comparing casein hydrolysate with a conventional CM-based formula. The ‘trial to reduce T1D in the genetically at risk’ is an international multicentre study with seventy-eight clinical centres in fifteen countries. The recruitment of families for the trial to reduce T1D in the genetically at risk study was completed at the end of 2006. Altogether, 2162 children were included in the intervention study and will be followed up until the age of 10 years.
BABYDIET
The German-wide BABYDIET study, an interventional trial, has been initiated to investigate whether delaying dietary gluten introduction influences the development of islet autoimmunity in newborns at genetically high risk for T1D and with a first-degree relative with T1D(Reference Schmid, Buuck and Knopff144). Children participating in BABYDIET are randomised to one of two dietary intervention groups that introduce gluten-containing cereals either at age 6 months, as recommended by the German National Committee for the Promotion of Breastfeeding, or at age 12 months (intervention group). The recruitment of children was finished in 2006 and altogether 150 children have been enrolled and will be followed up until the age of 10 years. The first results are expected for 2010.
The nutritional intervention to prevent type 1 diabetes pilot study
The nutritional intervention to prevent type 1 diabetes study has been initiated to investigate whether DHA supplementation during pregnancy and early childhood will prevent development of islet autoimmunity in children at high genetic risk for T1D and with a family history of T1D. Eligible participants (pregnant women or infants) will be randomised to one of the two study groups: a DHA group (intervention) or a control study group substance (placebo). During pregnancy and while breast-feeding, infants will receive the study substance indirectly through their mother (either via the placenta or via the breast milk). Infants who are either partially or exclusively formula fed will receive DHA more directly through the study formula. By 6–12 months of age, all the infants will get the supplement added to solid foods. Recruitment of families for the nutritional intervention to prevent study is still ongoing.
Maternal transfer of islet autoantibodies
The influence of maternally transmitted islet autoantibodies on the development of islet autoimmunity and T1D has been examined both in animal models and human subjects. In the non-obese diabetic (NOD) mouse, removal of maternally transmitted Ig prevented spontaneous diabetes in offspring mice, suggesting that maternal antibodies present during gestation including islet autoantibodies could be important factors in the pathogenesis of β-cell destruction(Reference Greeley, Katsumata and Yu145). Further studies in mice looking specifically at whether maternal insulin antibodies influence diabetes development reported controversial findings(Reference Koczwara, Ziegler and Bonifacio146, Reference Melanitou, Devendra and Liu147).
In the BABYDIAB study, 86 % of offspring from mothers with T1D have antibodies to exogenously administered insulin at birth and 66 % of offspring have antibodies to glutamic acid decarboxylase and/or islet antigens-2A at birth. The presence or absence of maternal insulin antibodies did not affect the risk of developing diabetes-associated autoantibodies and T1D in the child, but offspring with antibodies to glutamic acid decarboxylase and/or islet antigens-2A at birth had a significantly lower diabetes risk than offspring who were autoantibody-negative at birth(Reference Koczwara, Bonifacio and Ziegler148). Therefore, and in contrast to the data from animal studies, these findings in human subjects do not support the hypothesis that fetal exposure to islet autoantibodies increases the diabetes risk, but rather suggest that fetal exposure to antibodies to glutamic acid decarboxylase and/or islet antigens-2A may protect from future endogenous islet autoimmunity and T1D. Consistent with this observation is the overall decreased risk to develop islet autoimmunity and diabetes in offspring of mothers with T1D compared with that of offspring of fathers with T1D and nondiabetic mothers(Reference Warram, Krolewski and Gottlieb149, Reference Pociot, Norgaard and Hobolth150).
There is evidence that early exposure to environmental factors during pregnancy and/or early infancy influences the development of islet autoimmunity and T1D. However, the etiologic mechanisms that trigger autoimmunity and promote progression to disease are largely unknown. One major problem is that there is no access to the autoreactive T-cells within the pancreas that are responsible for the disease. Thus, we are not able to quantify and characterise these cells.
Advances in these areas are necessary if we want to fully understand the autoimmune pathogenesis of T1D. An international study (The Environmental Determinants of Diabetes in the Young), sponsored by the National Institutes of Health, is ongoing to address the early pathogenic mechanisms operating in islet autoimmunity(151). These are long but necessary studies that we hope will provide us with the knowledge of the environmental factors that affect the disease process.
Conclusions and future directions
The link between early dietary exposure and the onset of T1D in susceptible individuals remains unproven. However, most of the investigators in the area point to the increasing incidence of T1D as evidence that the most likely causative factor is environmental rather than genetic. In most of the cases, the results of studies that pinpoint a link between early diet and the later onset of T1D have been controversial and there are a few prospective case–control, cohort and human intervention studies that can be used for hypothesis testing. The conflicting evidence may, in part, be explained by the variation in the types of genetic predisposition and in the variable populations that have been used in studies. However, the major factor that prevents meaningful studies being carried out are the availability of biomarkers that allow researchers to detect the effects of the environment on autoimmunity and the likelihood of progression to the disease. It is the development of such environmentally responsive, pre-clinical biomarkers that will enable both mechanistic studies and improved prevention strategies to be developed. Many of the clinical advances in the treatment of T1D rely on post hoc interventions including islet transplantation, immune modulation and stem-cell therapy. The possibility of attempting to prevent the triggering of islet cell autodestruction or of minimising its effects until suitable therapy can be developed may act as a very useful adjunct to clinical intervention. It may also assist in preventing the onset of a second cycle of islet cell destruction after treatment.
Perinatal nutrition and neurodevelopment
Background to diet effects
Neuroanatomical and neurophysiological studies show that brain development occurs most rapidly during fetal development and in infancy. While family influences and the external environment undoubtedly play key roles in the child's cognitive development, there is evidence that there is a certain degree of stability in a childs' cognition which tracks from a very early age(Reference Bornstein, Hahn and Bell152). This suggests that early exposures possibly experienced in utero and postnatally may be involved in programming the brain and play a role in determining the cognitive ability.
Suboptimal cognitive ability is of considerable public health interest, because this leads to low educational attainment and these individuals tend to follow a trajectory of low socio-economic position, which is associated with fewer life chances and poorer adult health. In addition, both markers of restricted fetal growth and measures of cognition have been consistently associated with shorter life expectancy(Reference Batty, Deary and Gottfredson153) and increased prevalence of psychiatric outcomes such as depression(Reference Zammit, Allebeck and David154), schizophrenia(Reference Zammit, Allebeck and David154–Reference Osler, Lawlor and Nordentoft156) and suicide(Reference Mittendorfer-Rutz, Rasmussen and Wasserman157) as well as with adult chronic disease such as heart disease(Reference Batty and Deary158). A recent Scottish cohort study showed a 36 % increased risk of all-cause mortality per standard deviation decrease (15 points) in childhood intelligence quotient (IQ)(Reference Hart, Taylor and Smith159).
Epidemiological evidence for association
Evidence that cognitive ability may at least partly be determined early in life and may be related to nutrition comes from consistent associations between measures of fetal and infant growth and neurodevelopment, with low birth weight infants experiencing delays in reaching motor milestones and tending to have lower IQ(Reference Shenkin, Starr and Pattie160, Reference Richards, Hardy and Kuh161). In the ALSPAC, a population-based birth cohort of more than 14 000 children from the South West of England, who have been followed up for 18 years, birth length was associated with a decrease in the odds of having behavioural problems at 18 months of age(Reference Wiles, Peters and Heron162). A recent study suggested that, in adolescents assigned either a standard or a high-nutrient diet in the postnatal weeks after term birth, the high-nutrient group had significantly higher verbal IQ and caudate volume (as measured by magnetic resonance imaging). However, it should be noted that caudate volume correlated significantly with verbal IQ in the standard nutrient group only. The effect observed was selective to males only(Reference Isaacs, Gadian and Sabatini163).
Specific nutrients
With regard to which specific nutrients influence cognitive ability and behaviour, research has established that n-3 fatty acids, especially DHA which is found in abundance in the nervous system, are critical for infant growth and neurodevelopment. However, what is not clear is the effect of low levels of these nutrients on cognitive function. In ALSPAC, low seafood intake (known to contain high levels of n-3 fatty acids) by the mother during pregnancy was associated with an increased risk of suboptimal verbal IQ, prosocial behaviour, fine motor skills, communication skills and social development scores(Reference Hibbeln, Davis and Steer164). Furthermore, Rogers et al. (Reference Rogers, Emmett and Baker165) have shown that the frequency of IUGR in ALSPAC children decreased with increasing maternal fish intake – the OR of IUGR in those eating no fish was 1·85 (95 % CI 1·44, 2·38) compared with those in the highest fish intake group. Higher maternal intake of oily fish in ALSPAC has also been shown to be related to the offsprings' visual development(Reference Williams, Birch and Emmett166). Many other nutrients and micronutrients aside from n-3 fatty acids have been implicated in brain development and cognition. Iodine is an essential component of at least two thyroid hormones necessary for neurodevelopment, and iodine deficiency during pregnancy leads to fetal hyperthyroidism and irreversible neurological and cognitive deficits manifest as cretinism(Reference Black167). However, this may just be the tip of the iceberg, while most studies have looked at the effect of iodine supplements on cognition in socially deprived areas with low levels of iodine intake; a study carried out in 1221 school children in Spain, with iodine levels in the normal range, found that IQ was related to iodine intake(Reference Santiago-Fernandez, Torres-Barahona and Muela-Martinez168). A further study of 227 pregnant women in the North East of England found that approximately 40 % had borderline iodine deficiency(Reference Kibirige, Hutchison and Owen169). Another micronutrient that might influence cognition is Fe. Fe deficiency is the most common nutritional deficiency worldwide. A lack of sufficient Fe intake may significantly delay the development of the central nervous system because of alterations in morphology, neurochemistry and bioenergetics(Reference Beard170). Several observational studies have found that children who experience anaemia early in life continue along a trajectory of poor educational performance even after the anaemia had been treated. Other micronutrients which may be important in fetal brain development are B-vitamins, including folate(Reference Blaise, Nedelec and Schroeder171), choline(Reference Zeisel172), Zn(Reference Bhatnagar and Taneja173), vitamin D(Reference McGrath, Feron and Burne174) and cholesterol(Reference Herz and Chen175).
Critical developmental stage
Prenatal environment
A substantial body of work from animal models has demonstrated that imbalances in maternal nutrition during pregnancy can adversely affect normal fetal growth and neurodevelopment(Reference Perry, Kuh and Ben Shlomo176, Reference Lucas177). Indeed, aside from chronic exposure to tobacco smoke and alcohol, nutrition is probably the single greatest environmental influence both on the fetus and on the neonate, and plays a necessary role in the maturation and functional development of the central nervous system(Reference Morgane, Austin-LaFrance and Bronzino178). Inadequate nutrient availability during gestation may be related to psychiatric disease, behavioural problems, neurodevelopmental diseases, such as autism, and general cognition. In the human subjects, individuals exposed to famine in utero have increased risks of schizophrenia, showing long-term consequences of prenatal diet on the brain(Reference Susser, Hoek and Brown179, Reference McClellan, Susser and King180). Season of birth has also been shown to be associated with childhood IQ(Reference Lawlor, Ronalds and Clarke181), and nutrient availability could be responsible for this association. Nutrients found to be lacking in the mothers' diet could, if shown to be related to brain development, be modified to prevent disease and ensure that the child's cognitive ability is not impaired.
Postnatal environment
The strongest evidence for the effect of diet postnatally supports the developmental role of PUFA. Observational data from, for example, the Inuit in arctic Quebec(Reference Jacobson, Jacobson and Muckle182) show beneficial effects on development in general. This has also been supported by clinical studies on pre- and full-term infants(Reference Fleith and Clandinin183) and long-term studies on the effects of n-3 supplementation on visual and cognitive development throughout childhood(Reference Eilander, Hundscheid and Osendarp184).
Experimental evidence and mechanistic understanding
Developmental endpoints: intelligence quotient
A key problem is that observational studies of nutrition and IQ are subject to confounding by other lifestyle factors, which co-segregate with diet. This was highlighted in a recent large prospective cohort study which examined the association between breast-feeding and IQ; the authors found that before adjustment, breast-feeding was associated with an increase of about 4 IQ points, but adjustment for maternal intelligence accounted for most of this effect and when fully adjusted for a range of relevant confounders, the association disappeared(Reference Rankinen and Bouchard49). Measurement of diet is also problematic and often inaccurate due to wide exposure categories, misreporting of intake and recall bias(Reference Eilander, Hundscheid and Osendarp184). Large, well-conducted randomised control trials (RCT) are not subject to confounding and bias and a review of RCT of infants supplemented with n-3 fatty acids v. unsupplemented infants showed a positive effect on visual development, although evidence for neurodevelopment is inconsistent(Reference McCann and Ames185). However, most of these studies looked at infants' diet postnatally and not in utero. One small RCT of mothers' diet during pregnancy found that children who were born to mothers who had taken cod liver oil (n 48), which is high is n-3 fatty acids, scored higher on the Mental Processing Composite of the Kaufman Assessment Battery for Children at 4 years of age compared with children whose mothers had taken maize oil which is high in n-6 fatty acids (n 36)(Reference Helland, Smith and Saarem186). A study of Fe supplementation during pregnancy showed no effect on IQ at age 4, but supplementation only began at 20 weeks of pregnancy and a large proportion of children were lost to follow-up (30 %)(Reference Zhou, Gibson and Crowther187). Further trial evidence is needed, but such trials would have to recruit women before becoming pregnant in order to capture early gestation. An alternative study design would be to use a Mendelian randomisation approach(Reference Davey Smith and Ebrahim188, Reference Davey Smith and Ebrahim189). This could use the existing resources from large cohort studies to produce results more quickly and economically than an RCT and to identify more promising targets for RCT.
Developmental endpoints: others
Another factor that may also be relevant here in relation to cognitive, behavioural and eye development is the development of generalised movement during the first 6 months of life. This may provide a less confounded approach to map the formation of neural connections as a marker of brain development that can be determined relatively early in life, predicts neurodevelopmental outcome at 4 years and is influenced by LCPUFA status pre and postnatally in healthy term infants(Reference Bouwstra, Dijck-Brouwer and Wildeman190, Reference Smithers, Gibson and McPhee191).
Mendelian randomisation as a tool for overcoming confounding
Associations between genetic polymorphisms and phenotype are not generally subject to the problems of reverse causality, measurement error and confounding by lifestyle factors which occur in epidemiological studies(Reference Davey Smith and Ebrahim188, Reference Davey Smith and Ebrahim189, Reference Davey Smith and Ebrahim192). The use of genes as surrogates for measuring exposures in epidemiology has been termed Mendelian randomisation and is gaining recognition as an important research tool.
Genetic polymorphisms, which affect exposure to specific nutrients by influencing the diet, altering the metabolism or cell receptor function can be used to determine whether the related nutrients are important and to elucidate the important biological pathways. One example is the 5,10-methylenetetrahydrofolatereductase enzyme which controls a rate-limiting step in the folate metabolic pathway. The T allele at the C677T polymorphic site of this gene produces a thermolabile variant, which has a reduced catalytic capacity and results in less folate being available. This common genetic variant mimics the effect of low levels of folate in the diet, and associations have been found between this polymorphism in mothers and neural tubes defects(Reference Botto and Yang193), which are known to be caused by low levels of folate in the diet during pregnancy. Further exploitation of this concept could highlight the extent to which components of the mothers' diet influence neurodevelopment of the child.
Conclusions and future directions
There is substantial evidence, mainly from animal studies and observational data and associations from cohort studies, that the early environment and diet play a key role in neurodevelopment. In the prenatal and early neonatal period, the greatest environmental influence on neurodevelopment is most likely nutrition and most beneficially from breast milk. Dietary influences on cognitive development during the later postnatal period are more difficult to assess from observational studies, since such effects are subject to many confounders.
Among several micronutrients studied, DHA has shown to be critical for infant growth and neurodevelopment. Sub-optimal supply of other nutrients, e.g. iodine and Fe, to fetuses and neonates might affect a range of later physiological outcomes and, in particular, neurodevelopment(Reference Williams, Birch and Emmett166, Reference Kibirige, Hutchison and Owen169).
The influence of specific nutrients, in early life, on visual development has been extensively examined. Large well-conducted RCT have demonstrated a positive effect of n-3 fatty acids (i.e. DHA) supplementation on visual development in infants; however, for preterm infants, data are scarce, and the evidence is less strong. A recent Cochrane review(Reference Spittle, Orton and Doyle194) suggested that early intervention programmes for preterm infants appear to have a positive effect on cognitive outcomes in the short-to-medium term; however, further studies were recommended(Reference Bouwstra, Dijck-Brouwer and Wildeman190).
The use of validated early read-outs of cognitive, behavioural and visual development and the responsiveness of these to specific dietary interventions remain very relevant. General movement development during the first 6 months of life could be an important marker through which optimal neurodevelopment can be established(Reference Davey Smith and Ebrahim189, Reference Bouwstra, Dijck-Brouwer and Wildeman190).
Finally, Mendelian randomisation is gaining more recognition as being an important research tool by which existing resources from large cohort studies can be used. Genetic polymorphisms could be helpful to determine whether particular nutrients are potentially important and functional in the neurodevelopment of the child.
Perinatal nutrition and the atopic syndrome
Background to diet effects
The atopic syndrome manifests at barrier organs of the body to the environment. Therefore, clinical conditions on the skin (prototypic disease atopic eczema), the respiratory tract (allergic rhino-conjunctivitis and bronchial asthma) and at the mucosal site of the gastrointestinal tract (food allergies) play a major role. Despite many differences in the pathogenesis of these different diseases, there is a distinct pattern of common immuno-dysregulation(Reference Garn and Renz195–Reference Renz and Herz197). This comprises the development of chronic inflammatory disease starting out with a polarisation of T-cell effector responses towards a T-helper cell-2 phenotype. This T-helper cell-2 response controls many of the downstream effector mechanisms of allergic disease, including IgE antibody production, tissue and blood eosinophilia, mast cell activation and more.
Epidemiological evidence for association
Incidence and prevalence of atopic disease are still and constantly on the rise. Particularly, in Westernised countries, about one in three children are suffering from one type or another or a combination of the above-mentioned clinical phenotypes. This phenomenon clearly indicates that environmental factors play a decisive role in increasing the immunological susceptibility for the development of this immunological dysregulation(Reference von Mutius198, Reference von Mutius and Schmid199). Despite all advances in the development of anti-inflammatory medication, there is still no primary prevention available. Furthermore, the natural cause and chronology of the disease, as exemplified by airway remodelling in asthmatic patients, cannot be prevented or disrupted with present therapy. These aspects indicate the need for better therapy and preventive measures.
Critical developmental stage
Atopic children are born in a healthy state. The earliest clinical manifestation of the atopic syndrome manifests not before the first few months in life and are primarily located at the skin (atopic eczema) and the gut (food allergy). However, priming of adaptive immune responses occurs prenatally. It is now well established, for example, that antigen-specific T-cell of fetal origin is present in cord blood(Reference Prescott, Macaubas and Smallacombe200, Reference Szepfalusi, Nentwich and Gerstmayr201). This seems to be a physiological mechanism since virtually all newborns carry these antigen-specific immune responses. They are the result of intra-uterine antigen exposure and priming.
In the development of the immuno-pathogenesis of chronic inflammatory conditions including allergies and autoimmunity, the adaptive immune system plays a very prominent role. There are subtle differences between species in terms of the development of T- and B-cell responses within the prenatal environment. For example, in the human subjects, mature single CD4 or CD8 positive T-cells are readily detectable at about 17–20 weeks of gestation. As a consequence, antigens which pass the placental barrier can be presented to such mature and immature T-cells, leading to the development of specific T-cell immune responses. However, in mice, a species widely used for immunological research, such naïve mature T-cells, are present only 1–2 d before birth. Therefore, development of the antigen-specific T-cell repertoire mainly occurs under postnatal conditions in mice. Another example is the passage of maternal antibodies via the placental barrier. This is an active mechanism, at least partially relating to the expression of a unique type of Fcγ-receptors – namely FcγRn which bind, uptake and release maternal antibodies to the fetal site(Reference Uthoff, Spenner and Reckelkamm202, Reference Antohe, Radulescu and Gafencu203). The mechanism of pre and postnatal transfer varies for different Ig isotypes(Reference Gutierrez, Gentile and Miranda204, Reference Szepfalusi, Loibichler and Pichler205). Again, there are distinct species differences which must be considered if experimental studies are designed to further explore the immuno-regulatory mechanism at this time point.
However, there is no doubt that early programming contributes to a large extent for the development of a normal state of immunological responses. This normal state is characterised by the development of clinical tolerance against self-antigens as well as harmless environmental antigens. This level of clinical tolerance depends on T-cells, is antigen specific and must be acquired and maintained throughout life(Reference Renz and Herz197, Reference Breuer, Wittmann and Bosche206, Reference Bunikowski, Mielke and Skarabis207). Any interruption or disturbance of this physiological process will eventually lead to the development of disease. Concerning self-antigens, autoimmunity will occur; in the case of harmless environmental antigens, allergic and atopic disease will develop.
Experimental evidence and mechanistic understanding
Clinical endpoints can be considered as one important set of biomarkers and are useful to distinguish sub-phenotypes such as airway inflammation, airway hyperresponsiveness, development of IgE antibody profiles. Furthermore, clinical scores (SCORAD, the clinical scoring system for atopic dermatitis) have been established and shown to be useful in observational as well as interventional studies. Another set of biomarkers are immunological endpoints. They include phenotypes and markers of the innate as well as adaptive immune system. However, particular care must be taken in terms of quality control and establishment of age- and sex-specific normal ranges. An important aspect in this regard is the assurance of inter-laboratory and inter-centre-related quality control programmes.
Nutritional components and immune functions
There is abundant experimental as well as epidemiological evidence that many nutritional components are able to interfere directly or indirectly with certain immune functions. Prominent examples are the consumption of fish, fish oil and margarine containing more or less well-defined levels of n-3/n-6 fatty acids(Reference Waser, Michels and Bieli208). One important mode of action is via interference with the formation of phospholipids which particularly play an important role in the formation of arachidonic acid and its metabolites, including PG and leukotrienes which trigger and control certain inflammatory actions. Furthermore, obesity is to a certain degree linked to leptin levels, and, in turn, leptin itself has effects on immune functions(Reference Batra, Pietsch and Fedke209, Reference Fantuzzi, Sennello and Batra210). The same has been shown for vitamin D(Reference Froicu and Cantorna211). Another example for immune functions, particularly in the field of inflammation, is the balance of oxidative and anti-oxidative capacities(Reference Reichrath, Lehmann and Carlberg212). Again, this is influenced to some degree by nutritional factors, including vitamins and others.
Exposure to microbes or microbial components has also strong immuno-modulatory capacities. A prominent example in this area is the consumption of lactobacilli like Lactobacillus rhamnosus GG. Although the clinical benefits of prenatal and early postnatal Lactobacillus rhamnosus GG consumption are limited(Reference Blümer, Sel and Virna213, Reference Kalliomaki, Salminen and Poussa214), this approach clearly indicates that using microbes or microbial components opens an avenue for further exploration. This is also highlighted by a number of experimental studies indicating strong in vitro or in vivo (animal model systems) effects on the development of innate and adaptive immune responses(Reference Renz and Herz197). In addition, prebiotics that fuel the intestinal microflora can also affect the development of the immune system(Reference Boehm, Jelinek and Knol215).
Although we have gained great insights into underlying mechanisms of nutritional and microbial immuno-modulation, particularly at early time points, we are still some way from fully understanding the effects of such interference in detail. However, such understanding is necessary to fully apply this intriguing concept to allergy prevention in human subjects. Therefore, suitable model systems must be developed for further exploration into the mechanistic fundamentals of this approach. In this regard, we have made some progress recently by designing murine models for various allergic phenotypes including acute and chronic experimental asthma with the development of airway remodelling(Reference Wegmann, Fehrenbach and Fehrenbach216). These disease-related models emulate more closely the real situation in patients compared with the previous ones.
Conclusion and future directions
Development of prevention and improved intervention strategies is the major goal in the area of allergy and asthma research. The intriguing advantage of using nutritional and microbial components is the ease of safe application, particularly if applied during pregnancy, as clinical effectiveness must be paired with safety. Furthermore, the benefit of this intervention/prevention must be long lasting and causes no impairment of other types of immune responses. To develop a nutritional solution, which fulfils all of these criteria, is a major research challenge and to reach this aim, different avenues need to be explored. One approach is the molecular analysis of nutritional factors, which are associated with a reduced risk for the development of allergic disease. Another level of investigation is the establishment of a proof of concept in suitable animal models mimicking the human phenotype as closely as possible. Furthermore, it will be very important to get detailed information about the underlying mechanisms since this will result in further improvement of the preventive approach. Finally, clinical studies are required to prove the effectiveness of this approach, particularly under long-term conditions. To reach this goal, an interdisciplinary network must be established, combining the expertise of epidemiologists and clinicians, together with basic scientists from the fields of cellular biology, immunology, biochemistry and molecular biology.
Perinatal nutrition and bone health in adults
Background to diet effects
Osteoporosis is a major and increasing cause of morbidity and mortality in developed countries, and set to become worldwide in the next few decades. The cost of treating fragility fractures in the UK is £1·73 billion/year(Reference Torgerson, Barlow, Francis and Miles217), close to that for treating CVD (£1·75 billion)(218). The possibility that nutritional interventions in infancy could reduce the burden of adult degenerative bone disease is therefore an important public health issue(Reference Winsloe, Earl and Dennison219). This section of the review will address three questions:
(1) What are suitable measures or biomarkers of bone health in human subjects?
(2) How well do these measures predict later outcome?
(3) What are the key early factors that influence later bone health and can the effects of adverse early factors be overcome later in life?
Measures or biomarkers of bone health in human subjects
The ideal outcome measure, osteoporotic fracture, is for obvious reasons rarely available, and bone biopsies cannot be obtained in healthy children, precluding the use of histological or histomorphometric measures. In practice, therefore, investigators are reliant on proxy measures that can be easily obtained in healthy infants and children.
Bone mass or density (BMD) is generally obtained using Dual X-ray Absorptiometry (DXA). In postmenopausal women, DXA BMD is a significant predictor of a clinical outcome, i.e. fracture risk. However, the predictive value of BMD in children is much less clear and it cannot be automatically assumed that BMD is the optimal DXA-derived parameter to use in studies examining the effects of early life factors on later bone health(Reference Tobias, Steer and Emmett220).
Quantitative computed tomography is used to make structural bone measurements, including volumetric bone density of the peripheral skeleton (tibia and radius), quantitative computed tomography is the best compromise between the need for more detailed measures yet still with minimum radiation exposure. Radial quantitative ultrasound measurements predict fracture risk in adults(Reference Nguyen, Center and Eisman221), independently of BMD, and may reflect aspects of bone structure not captured by DXA.
Bone is a dynamic tissue, constantly undergoing remodelling, in which resorption is followed by bone formation at the same site, allowing bone to adapt to biomechanical stresses and for old or damaged bone to be replaced. Growing bone also undergoes modelling, in which formation and resorption are uncoupled and occur at different sites, with resorption at endosteal surfaces and formation at periosteal surfaces. Bone formation and resorption can be measured using a variety of specific markers which are released into blood or urine. Markers of bone formation include osteocalcin, bone-specific alkaline phosphatase and amino-terminal procollagen propeptides of type I collagen, released at different stages of osteoblast proliferation and differentiation(Reference Rauchenzauner, Schmid and Heinz-Erian222). Markers for bone resorption are degradation products of type 1 collagen that can be quantified in blood – plasma carboxyterminal telopeptide 1 chain of type I collagen (CTX) – or urine – N-telopeptides of type-I collagen (NTX) and deoxypyridinoline normalised to creatinine. In adult populations, and in some paediatric diseases, these markers can be useful clinical tools for monitoring the response to treatment. However, levels of bone turnover markers are influenced by many factors, including age, sex, time of day, season and pubertal stage, which makes interpretation particularly difficult in children. Furthermore, different methods and assay kits produce different values for the same marker and cannot be used interchangeably. Bone turnover markers provide a qualitative assessment of bone metabolism and may be informative when longitudinal measurements are made under standardised conditions or when comparisons can be made between randomised groups. They typically correlate poorly with measurements of bone mass in children.
Epidemiological evidence for association
Bone mass measurements during childhood have been shown to predict fracture risk over the subsequent 2 years(Reference Clark, Ness and Bishop223) or 4 years(Reference Goulding, Jones and Taylor224). Not surprisingly, there are no longitudinal studies relating measurements in childhood with outcome in the same individual. Nevertheless, peak bone mass is generally accepted to be a good predictor of osteoporosis risk. Using computer modelling, Hernandez et al. (Reference Hernandez, Beaupré and Carter225) predicted that a 10 % increase in BMD would delay the development of osteoporosis (defined as BMD < 2·5 sd from the young adult mean) by 13 years, whereas a similar change in age at menopause or non-menopausal bone loss would only result in a delay of 2 years. There are no reliable data on the predictive value of quantitative computed tomography measurements in childhood for later outcome, but it seems reasonable to extrapolate the likely effects of observed changes in bone geometry (especially those seen in later childhood or adolescence) on bone strength to effects in later life. In contrast, it is more difficult to predict the consequences of differences in bone turnover markers in childhood for later bone health.
Critical developmental stage
A number of factors have been shown to result in increased bone mass in the short term, during the period of intervention. This may in itself have immediate outcome benefits for the individual; for example, reducing short-term fracture risk. However, to represent a potential preventative strategy against osteoporosis, any such effect must be shown to persist after the intervention has stopped, resulting in higher peak bone mass and/or favourable effects on bone structure or bone turnover. This has received much less attention.
Studies in human subjects suggest that influences in fetal life, infancy and possibly childhood may programme skeletal growth trajectory and later bone health. Data from the Southampton Women's Study suggest lower maternal fat stores, vigorous activity in late pregnancy, maternal smoking and low maternal birth weight, all predict lower neonatal bone mass(Reference Godfrey, Walker-Bone and Robinson226). Mechanistic explorations suggest the association between maternal fat stores and infant bone mass can be explained by umbilical venous leptin(Reference Javaid, Godfrey and Taylor227). Both cord leptin and IGF-1(Reference Javaid, Godfrey and Taylor228) closely predict neonatal skeletal size. Maternal vitamin D insufficiency or deficiency (seen in 49 % of women) during late pregnancy was also associated with reduced bone size and mineral mass in the offspring at 9 years(Reference Javaid, Crozier and Harvey9).
Analyses in both historical and modern prospective cohorts have shown that birth weight is positively associated with later bone mass, via an effect on body and skeletal size(Reference Cooper, Fall and Egger229–Reference Fewtrell, Prentice and Cole232) and that more rapid growth during infancy and childhood is associated with higher bone mass in later life(Reference Fall, Hindmarsh and Dennison233, Reference Weiler, Yuen and Seshia234). Weight in infancy predicts adult bone mass independently of adult lifestyle, possibly by programming of the IGF-1/growth hormone axis(Reference Weiler, Yuen and Seshia234). Importantly, data from retrospective historical cohorts suggest that differences in weight at 1 year of age predict differences in proximal femoral geometry (an independent predictor of hip strength and fracture risk) in later adult life(Reference Javaid, Lekamwasam and Clark235) and that increased linear growth during childhood predicts a lower risk of osteoporotic fracture(Reference Cooper, Eriksson and Forsen236).
Experimental evidence and mechanistic understanding
That infant nutrition could influence later bone health has also been the subject of several studies. Breast-feeding was associated with higher bone mass in children born at term(Reference Jones and Dwyer231) and our(Reference Fewtrell, Williams and Singhal253) data suggest a similar beneficial effect of human milk on peak bone mass in subjects born preterm. In our experimental studies, in infants randomly assigned to diet during early postnatal life, children born preterm and randomised to lower nutrient diets showed biochemical evidence of increased bone formation later in childhood(Reference Fewtrell, Prentice and Jones237). In the same cohort, those who developed (usually silent) metabolic bone disease due to inadequate early intake of Ca and P were shorter at 8–12 years, suggesting adverse programming of linear growth(Reference Fewtrell, Cole and Bishop238). Conversely, nutritional interventions later in childhood have less convincing long-term effects. While Ca supplementation may have short-term benefits for bone mass, these are generally lost once the intervention is withdrawn, and there is little evidence for a clinically relevant persisting effect of childhood Ca supplementation on long-term bone health(Reference Winzenberg, Shaw and Fryer239). There are theoretical reasons why other elements of the diet such as vitamin K, Zn, protein, Na or fruit and vegetables might influence later bone health, but very few studies have yet been conducted on specific nutrients.
Weight-bearing physical activity has attracted increasing interest as a potential modifiable determinant of peak bone mass. A number of randomised intervention studies in children and adolescents have demonstrated increased bone mass in loaded bones during the period of increased activity. Collectively, the results of these studies suggest that effects are site specific, greatest for cortical bone and that interventions may be most effective during puberty when bone growth is most rapid. Although the majority of studies have used DXA to measure bone mass, some have also reported higher cortical cross-sectional area, cortical thickness and increased parameters of bending strength, suggesting that weight-bearing exercise may have benefits for bone structure and bone mass. Follow-up of individuals who have participated in intervention trials is limited but two studies have demonstrated effects persisting after the intervention ceased on hip bone area and bone mineral content(Reference Fuchs and Snow240) and tibial periosteal circumference(Reference Specker, Binkley and Fahrenwald241). In some studies, the effect of weight-bearing exercise was seen only in subjects with the highest Ca intakes(Reference Specker, Binkley and Fahrenwald241, Reference Iuliano-Burns, Saxon and Naughton242).
Conclusion and future directions
In conclusion, despite limitations in the range of available measures for assessing later bone health, there is evidence that osteoporosis risk may be at least partly modified by interventions during early life designed to optimise linear growth, nutrition and weight-bearing activity. The ‘critical period’ during which bone health can be programmed may well extend throughout childhood and adolescence while the skeleton is still growing. It is relevant to consider the likely practical relevance of the observed effect sizes. Later bone mass in subjects who received breast milk was about 0·4 sd higher than in those who received formula(Reference Jones and Dwyer231) – about 12 % of the population variance. The effects of weight-bearing exercise interventions on BMD are in the order of 3–5 %. While there are difficulties and uncertainties inherent in extrapolating bone mass data from children to adults, it has been calculated that a 2–3 % increase in peak bone mass could reduce later fracture risk by 10–20 %. Hence, available data suggest that the effect sizes observed with early interventions may be of a magnitude which could be potentially significant in public health terms in reducing the burden of osteoporosis.
Conclusion
Metabolic imprinting/programming is an increasingly important concept that may prove to be the single most important mode of successful dietary intervention to improve health. While much of the effort has been concentrated towards early life (pre and early postnatal) as the most significant developmental stages, there is some evidence that, for certain health endpoints, a longer/later intervention may also be successful.
The key opportunities for interventions, which have been outlined in this review, include obesity, CVD, bone health, cognition, immune function and diabetes. These are health endpoints for which observational/epidemiological evidence for programming exists. Underpinning many of these and taking them beyond epidemiology and observational studies are the experimental intervention investigations that make use of both human and animal subjects. Providing a mechanistic basis for many of the observed effects are the epigenetic studies looking at specific molecular events that extend beyond genetic polymorphisms and provide a programmable and exquisite way of controlling gene expression and subsequent phenotype.
There remains considerable gaps in the knowledge and challenges, not least in the development of suitable systems to test hypotheses but the ultimate goal will be in the controlled and predictable beneficial manipulation of human health at all life stages in the short-term dietary interventions leading to long-term improvements in health.
Acknowledgements
The present article has been written to reflect the presentations and discussions from the Workshop on Mechanisms and Definitions of Metabolic Imprinting, Programming and Epigenetics, organised on 5–6 June 2007 in Florence, Italy. Each author provided the scientific content for his or her respective chapter, which reflects their contribution to the workshop. B. H. is employed by Wrigleys, M. G. by FrieslandCampina, L. H. by Mead Johnson Nutrition, K. M. by Nestlé and Dr v. D. B. by Danone. No other conflicts of interest have been declared. The work was commissioned and funded by the Metabolic Imprinting Task Force of the European branch of the International Life Sciences Institute (ILSI, Europe). Industry members of this task force are Danone, FrieslandCampina, Martek Biosciences Corporation, Mead Johnson Nutrition and Nestlé. For further information about ILSI Europe, please email info@ilsieurope.be or call+32 2 771 00 14. The opinions expressed herein are those of the authors and do not necessarily represent the views of ILSI Europe.





