


Since the 1980s, studies that have examined the effects of maternal nutrition during pregnancy on the risk of degenerative diseases later in life have indicated that fetal development can be affected by maternal over- and undernourishment( Reference Barker, Osmond and Law 1 – Reference Vickers, Breier and Cutfield 5 ). The most famous researchers in this field are Barker et al. ( Reference Barker, Osmond and Law 1 ), who studied the association between the mortality rates of CVD and neonatal mortality more than 70 years ago in different areas of England and Wales. They suggested that poor nutrition in early life increased susceptibility to CVD( Reference Barker 2 ). Population cohort studies have indicated that adults who were exposed to the Dutch famine (which occurred during the winter of 1944–5) in utero tended to have an atherogenic lipid profile( Reference Roseboom, van der Meulen and Osmond 6 ), high BMI, large waist circumference( Reference Ravelli, van Der Meulen and Osmond 7 ) and high risk of CHD( Reference Roseboom, van der Meulen and Osmond 8 ). These studies gave evidence that showed an association between nutritional experience in early life and the risk of degenerative disease in later life. They also gave clear evidence that degenerative diseases, such as CHD, hypertension, dyslipidaemia, obesity and diabetes, could be initiated in utero.
Several animal models have been developed to investigate the molecular mechanism. Researchers have designed several diets, including energy-rich diets( Reference Williams, Seki and Vuguin 9 ), protein-restrictive diets( Reference Altobelli, Bogdarina and Stupka 10 ) and micronutrient-deficient diets( Reference Tomat, Costa Mde and Arranz 11 ), to feed pregnant and/or lactational rodents in order to study the effects of maternal diet on the health status of offspring in later life. Some studies( Reference Melo, Benatti and Ignacio-Souza 12 , Reference Benatti, Melo and Borges 13 ) have shown metabolic disorder in the offspring of dams that were fed a high-lipid, high-energy (HLE) diet during gestation and lactation; however, the specific molecular mechanisms underlying these changes were not very clear. Currently, the most famous mechanism is epigenetics( Reference Osborne-Majnik, Fu and Lane 14 ). In 1998, Wolff et al. ( Reference Wolff, Kodell and Moore 15 ) reported that feeding pregnant black a/a dams methyl-supplemented diets (supplemented with folic acid, vitamin B12, betaine and choline) changed the epigenetic regulation of agouti expression in their offspring. That study indicated that the maternal diet is important for the epigenetics of offspring. Some researchers have tested this hypothesis and have found that altering the maternal diet, e.g. by introducing a protein-deficient diet( Reference Altobelli, Bogdarina and Stupka 10 , Reference Rees, Hay and Brown 16 ) or a micronutrient-deficient diet( Reference Wolff, Kodell and Moore 15 ), changed the DNA methylation profile in offspring. Recently, Cannon et al. ( Reference Cannon, Buchner and Hester 17 ) reported that maternal diet has a pervasive effect on gene expression, but transcriptional changes are unlikely to be caused by DNA methylation differences. However, Marco et al. ( Reference Marco, Kisliouk and Tabachnik 18 ) reported that even when offspring from dams that were fed HLE diets during gestation and lactation ate chow diet (CD) from weaning, the offspring displayed increased Pomc promoter hypermethylation. This discrimination was limited by sample size, body weight before gestation, diet composition and other factors.
In our earlier study( Reference Yu, Miao and Gao 19 ), we reported that global gene expression decreased in neonatal mice livers in order to adapt to a maternal HLE diet during gestation and lactation. It has been suggested that the maternal consumption of a high-fat diet can change the epigenetic marks in offspring and alter gene expression in the long term( Reference Vucetic, Kimmel and Totoki 20 ). In the present study, we used microarrays to analyse the global methylation changes in gene promoter sites in order to find the mechanism that underlies the global decreasing gene expression that we observed in our earlier study.
Material and method
Animals and diets
The Beijing Administrative Committee for Laboratory Animals and the Ethical Committee for Animal Care and Use of the Capital Medical University reviewed and approved all animal experiments carried out in the present study.
Male and female C57BL/6J mice (specific pathogen free, 9 weeks old) were housed in groups of four mice per cage at 22°C with a 12 h light–12 h dark cycle, and they were given free access to food and water. After 1 week, fifteen male and fifteen female mice were transferred to one cage and fed either a control CD or an HLE diet (composed of 84 % CD+15·8 % lard fat+0·2 % cholesterol). These same diets were used in one of our previous studies( Reference Cannon, Buchner and Hester 17 ). Total food intake and body weight gain were recorded twice a week. There were ten pregnant CD dams and ten pregnant HLE dams that gave birth to litters ranging from six to ten pups. One male pup from each dam was randomly assigned to the present experiment. After 21 d, all of the weaning offspring (n 10 in each group) were fed the CD until they were 10 weeks old, at which time they were killed by cervical vertebra dislocation. Livers were sampled, immediately frozen in liquid N2 and stored at − 80°C until further study.
Genomic DNA, total RNA, and protein isolation from livers
Total RNA, genomic DNA and proteins were purified by commercial kits (GenElute RNA/DNA/Protein Plus Purification Kit, catalogue number E5163) provided by Sigma. All of the procedures were conducted according to the manufacturer's instructions. In brief, 20 mg liver tissues from one of the twenty mice were minced thoroughly using a pestle and transferred into a DNA purification column. After spinning and washing repeatedly, the genomic DNA, total RNA and protein were separated and purified.
Global DNA methylation quantification
The methylation of the genomic DNA was detected using the Methylamp global DNA methylation quantification ultra kit (Epigentek) according to the manufacturer's operational guide. In this assay, genomic DNA is immobilised in strip wells specifically coated with a DNA-affinity substance. The methylated fraction of DNA can be recognised by a 5-methylcytosine antibody and quantified through an ELISA-like reaction. The amount of methylated DNA is proportional to the optical density (OD) intensity. Absorbance of positive (methylated) and negative (unmethylated) control DNA were measured by a microplate reader (Tecan). The amount of DNA methylation was calculated using the following formula:
 $$\begin{eqnarray} Methylation\,\% = \frac {(sample\,OD - negative\,control)/ X ^\ast }{(positive\,control\,OD - negative\,control\,OD)\times 10}\times 100\,\%. \end{eqnarray}$$
$$\begin{eqnarray} Methylation\,\% = \frac {(sample\,OD - negative\,control)/ X ^\ast }{(positive\,control\,OD - negative\,control\,OD)\times 10}\times 100\,\%. \end{eqnarray}$$
The amount of the positive control was 10 ng, and sample DNA was adjusted to 100 ng. X* is the GC content of the species DNA; in the present study, it is 42 %.
Microarray analysis of DNA methylation and data analysis
The Mouse Promoter 1.0R Array (Affymetrix) was used to detect DNA methylation. It is a single array comprised of more than 4·6 million probes that are tiled to interrogate more than 28 000 mouse promoter regions. The entire procedure was conducted according to the manufacturer's instructions. In brief, genomic DNA was purified using a kit (Qiagen) on three samples from each group. Each sample included a mixture of DNA from three mice livers that were randomly selected from the ten livers. Methylated DNA was gathered using the EpiQuik Methylated DNA Immunoprecipitation Kit (Epigentek). Target DNA was amplified, fragmented and labelled. After hybridisation, washing and staining, the chip was scanned according to the manufacturer's instructions. First, all of the locations where the degree of methylation was significantly changed were identified, and the gene names were annotated. We applied a random variance model (RVM) t test to filter the differentially expressed methylation peak genes for the control and experiment groups, because an RVM t test can raise df effectively in the case of small samples. After the significant analysis and false discovery rate analysis, we selected the differentially expressed genes according to the P value threshold( Reference Wright and Simon 21 – Reference Clarke, Ressom and Wang 23 ).
Then, all of the genes that had been identified by microarrays as being significantly hypermethylated were analysed using gene ontology (GO) analysis and pathway analysis, which were outlined in a study that we published earlier( Reference Yu, Miao and Gao 19 ).
Gene expression analysis by real-time PCR
The total RNA that had been purified earlier was reverse transcribed using an RT kit (no. A3500; Applied Promega). The mRNA encoding PPARγ, liver X receptor α (LXRα), cluster of differentiation 36 (CD36) and β-actin (which was used as an invariant control) were analysed by real-time PCR. The primer sequences are listed in Table 1. PCR was carried out as follows: denaturation at 94°C for 5 min for the first cycle, and then cycles consisting of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s.
Table 1 Primers sequence used in the present study

F, forward; R, reverse; LXRα, liver X receptor α; CD36, cluster of differentiation.
Western blot analysis
For the Western blot analysis, livers from adult mice were homogenised on ice in radioimmunoprecipitation assay (RIPA) lysis buffer (Sigma) containing phenylmethanesulfonyl fluoride as a proteinase inhibitor. Homogenates were centrifuged for 10 min at 12 000 rpm at 4°C, and the supernatants were collected. The total protein content was determined using the bicinchoninic acid method. Then, 50 μg of protein extracts from each liver tissue were resuspended in an SDS-containing buffer, maintained in boiling water for 5 min and separated by SDS-PAGE. After electrophoresis, proteins were transferred to nitrocellulose membranes, and PPARγ and LXRα were detected using an anti-PPARγ antibody (ab3259, 1:1000) and an anti-LXRα antibody (ab58092, 1:1000). An anti-β-actin antibody (ab, 1:1000) was used as a control. Signals were revealed using an enhanced chemiluminescence (ECL) kit (Fivephoton Biochemicals), and the dilution ratio of IgG was 1:5000. ImageJ was used to analyse the density of the bands.
Statistical analysis
Results are expressed as means with their standard errors. SPSS (version 11.5; SPSS Institute, Inc.) was used for statistical analysis. Significant differences were assessed using Student's t test (two-tailed). A P value of < 0·05 was considered statistically significant.
Results
The genomic DNA in adult mice livers was hypermethylated by a maternal high-lipid, high-energy diet during pregnancy and lactation
The global DNA methylation degree was measured using commercial kits. The results indicated that the percentage of methylated DNA in mice livers from offspring delivered by dams that were fed an HLE diet during pregnancy and lactation was significantly higher than that in the CD group (Fig. 1). The percentage of methylated DNA in the CD group (n 10) was 24·55 %, whereas it was 29·24 % in the HLE group (n 10).
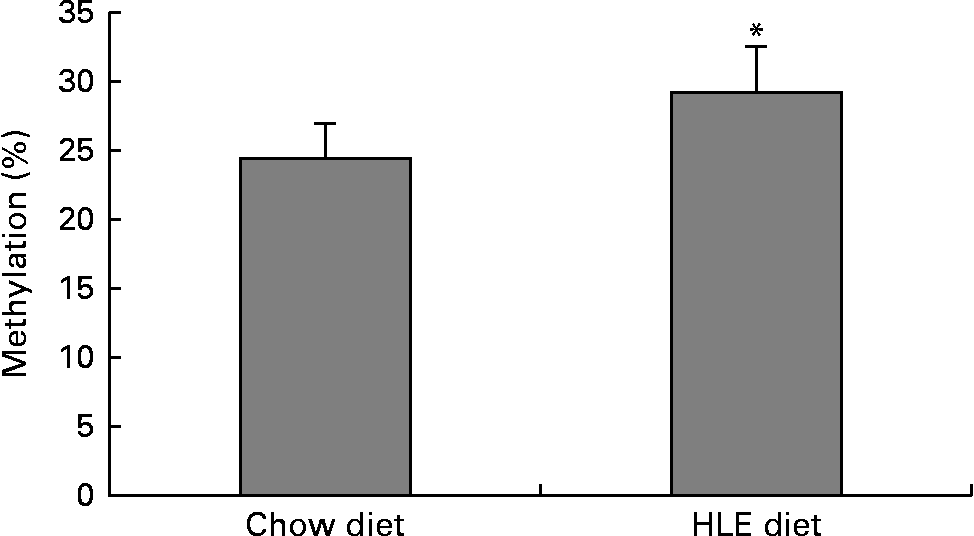
Fig. 1 The amount of methylated DNA in the livers of adult male offspring from dams that were fed a chow diet (CD) or a high-lipid, high-energy (HLE) diet during pregnancy and lactation. Genomic DNA from mice liver was extracted, and the methylated DNA was captured by a 5-methylcytosine antibody and quantified through an ELISA-like reaction. Values are means (n 10), with their standard errors represented by vertical bars. * Mean value was significantly different compared to that in the CD group (P≤ 0·05).
The hypermethylated genes in adult mice livers were identified by microarray
There were 1635 genes identified as being significantly hypermethylated by a maternal HLE diet (online supplementary Table S1), including some famous genes related to glucose, fatty acids and cholesterol metabolism, such as PPARγ, nuclear receptor subfamily 1, group H, member 3 (NR1H3) (LXRα), Insig2 (insulin induced gene 2), CD36 and Pcsk9 (proprotein convertase subtilisin/kexin type 9) (refer to online supplementary Table S1). All of these hypermethylated genes were distributed in the nineteen chromosomes shown in Fig. 2.

Fig. 2 The localisation of hypermethylated genes in chromosomes (chr) recognised by the mouse promoter 1.0R Array provided by Affymetrix. ![]() , Hypermethylation;
, Hypermethylation; ![]() , centromere. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
, centromere. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
Bioinformatic analysis mapped the relationship between the functions of the genes which were hypermethylated
Then, all of the genes that had been identified as being significantly hypermethylated were analysed using GO analysis and pathway analysis, which were described in a study that we published earlier( Reference Yu, Miao and Gao 19 ). GO analysis indicated that there were 261 GO (online supplementary Table S2) significantly affected by the maternal diet. GO map analysis helped determine the relationship between the differentially expressed and upstream genes. In this map, a total of twenty-four groups of functions were affected, including phosphorylation protein (GO:0006468), the Wnt receptor signalling pathway (GO:0016055), the regulation of the apoptotic process (GO:0042981) and the apoptotic process (GO:0006915) etc. (Fig. 3).

Fig. 3 Gene ontology (GO) map made from significant GO regulated by a maternal high-lipid, high-energy (HLE) diet. Based on the GO analysis, significant GO affected by a maternal HLE diet during gestation and lactation were selected, and the GO map was made. All of these GO were down-regulated. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
Functions related to lipid storage (GO:0019915), the lipid metabolic process (GO:0006629), the negative regulation of LDL particle clearance (GO:0010989), cholesterol homeostasis (GO:0042632), the fatty acid biosynthetic process (GO:0006633), the long-chain fatty acid metabolic process (GO:0001676), the sphingolipid metabolic process (GO:0006665), the regulation of the steroid metabolic process (GO:0019218), the regulation of cholesterol transport (GO:0032374), the regulation of the LDL particle receptor catabolic process (GO:0032803), the long-chain fatty-acyl-CoA metabolic process (GO:0035336), multicellular organismal lipid catabolism (GO:0044240), LDL particle mediated signalling (GO:0055096), the response to LDL particle stimulus (GO:0055098), the lipid biosynthetic process (GO:0008610), the TAG metabolic process (GO:0006641), the saturated monocarboxylic acid metabolic process (GO:0032788), the unsaturated monocarboxylic acid metabolic process (GO:0032789) and lipid transport (GO:0006869) were also significantly affected.
Pathway analysis found that there were fifty-five pathways (online supplementary Table S3) that were affected by the maternal diet, including metabolic pathways, pathways in cancer, the mitogen-activated protein kinase signalling pathway, the Wnt signalling pathway and the Ca signalling pathway etc. The insulin signalling pathway (pathway ID: 4910), the biosynthesis of unsaturated fatty acid pathway (pathway ID: 1040) and the PPAR signalling pathway (pathway ID: 3320) were also significantly affected by the maternal diet. Some of these pathways interacted with each other, as was revealed by the pathway net analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Fig. 4). The analysis indicated that the insulin signalling pathway was an upstream pathway, and changes to it resulted in changes in other pathways.

Fig. 4 Pathway net constructed from significant pathways regulated by a maternal high-lipid, high-energy (HLE) diet during gestation and lactation. Based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, fifty-five significantly changed pathways were identified, and a pathway network was created. MAPK, mitogen-activated protein kinase. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
The expression of genes related to lipid metabolism were inhibited by a maternal high-lipid, high-energy diet as measured by real-time PCR and Western blot
The PPARγ, LXRα and CD36 genes, which are related to lipid metabolism in hepatic tissues of adult offspring, were found by microarray to be hypermethylated. DNA methylation leads to the inhibition of gene transcription. The mRNA and protein expression of these genes were therefore measured by real-time PCR and Western blot analysis. The results indicated that PPARγ and LXRα mRNA and protein expression were significantly decreased in the HLE group (Fig. 5(a) and (b)). The CD36 mRNA expression was also significantly inhibited by a maternal HLE diet (Fig. 5(a)).

Fig. 5 Expression levels of mRNA and protein in livers of male adult offspring from dams fed a chow diet (CD) or high-lipid, high-energy (HLE) diet. (a) The mRNA levels of PPARγ, LXRα (liver X receptor α) and CD36 (cluster of differentiation 36) in male adult mice livers measured by real-time PCR (n 7). ![]() , CD;
, CD; ![]() , HLE diet. (b) The nuclear PPARγ and LXRα protein content in male adult mice livers measured by Western blot (n 6).
, HLE diet. (b) The nuclear PPARγ and LXRα protein content in male adult mice livers measured by Western blot (n 6). ![]() , CD;
, CD; ![]() , HLE diet. Values are means, with their standard errors represented by vertical bars. * Mean value was significantly different compared to CD (P≤ 0·05).
, HLE diet. Values are means, with their standard errors represented by vertical bars. * Mean value was significantly different compared to CD (P≤ 0·05).
Discussion
It has been reported that a maternal HLE diet during pregnancy induces the global inhibition of gene expression in neonatal mice livers, and lower levels of the expression of these genes remained in adulthood when offspring were fed CD after weaning( Reference Yu, Miao and Gao 19 ). We hypothesised that there might be global gene promoter hypermethylation in the liver. Using the Mouse Promoter 1.0R Array, we carried out a genome-wide analysis of the DNA methylation of gene promoters in adult mouse livers to verify this hypothesis. The results indicated that the promoter hypermethylation of genes existed, and some of these genes were involved in the lipid metabolism and pathways.
Intra-uterine life is a complex developmental programme that involves cell division, growth and differentiation( Reference Bird 24 ). The ability to sense, interpret and react to environmental stimuli by changing gene expression is a basic survival skill of cells. This process includes the modification of DNA by methylation at CpG sites( Reference Jaenisch and Bird 25 ). Nutrients transferred from dams to fetuses are important environmental stimuli. It has been reported that a maternal low-protein diet significantly increases the methylation of PPARα in offspring livers( Reference Altmann, Murani and Schwerin 26 ). In another study, global and gene-specific (dopamine reuptake transporter, μ-opioid receptor and preproenkephalin) promoter DNA hypomethylation were observed in the brains of offspring from dams that were fed a high-fat diet( Reference Marco, Kisliouk and Tabachnik 18 ). In oocytes of obese mice that were induced by a high-fat diet, the leptin promoter DNA was significantly hypermethylated, but the PPARα was hypomethylated( Reference Ge, Luo and Lin 27 ). This trend was also observed in the livers of female offspring from dams that were fed a high-fat diet( Reference Ge, Luo and Lin 27 ). These results indicated that the epigenetic mechanism underlies the effects of maternal diets on gene expression in offspring. However, there is significant discrepancy. Recently, a study reported that a maternal high-fat diet had a pervasive effect on gene expression but not on DNA methylation in the liver( Reference Cannon, Buchner and Hester 17 ), but another research group reported that offspring from dams that were fed an HLE diet during gestation and lactation displayed increased hypermethylation at the promoter of proopiomelanocortin gene( Reference Marco, Kisliouk and Tabachnik 18 ).
In the present study, the specific genes whose promoter DNA were significantly hypermethylated or hypomethylated were scanned using microarray on liver tissue. Among the 1635 genes, only seventeen genes were hypomethylated significantly, while the others were significantly hypermethylated in the livers of offspring from dams that were fed an HLE diet. Because of the absolute advantage of the hypermethylation of genes, the results of the global DNA methylation measurements indicated that the percentage of methylated DNA in livers in the HLE group was significantly higher than that in the CD group. The significant heterogeneity of factors such as the fatty acids category in the diet, the genes that were selected to be observed and the number of dams and offspring used in the studies( Reference Cannon, Buchner and Hester 17 , Reference Llopis, Sánchez and Priego 28 ) made inconsistent results between the studies predictable.
Many studies found down-regulation in some important genes that regulate lipid and glucose metabolism. In Laker et al.'s study( Reference Laker, Lillard and Okutsu 29 ), the mRNA expression of PPARγ co-activator-1α (Pgc-1α) and its target genes were inhibited in the muscular tissue of neonatal and adult mice from dams that were fed a high-fat diet during gestation and lactation. Magliano et al. ( Reference Magliano, Bargut and de Carvalho 30 ) found that a maternal pre-gestational and gestational high-fat diet diminished the expression of PPARα in the livers and PPARγ in the white adipose tissue of adult offspring. In the present study, the nuclear protein levels of PPARγ and LXRα were significantly decreased in the livers of mice that were the offspring of dams that were fed an HLE diet. Both PPARγ and LXRα were also suggested as hypermethylated genes by microarray measurement.
PPARγ was found predominantly in fat tissues and to a lesser extent in other tissues, including the liver( Reference Tsai and Maeda 31 ). PPARγ plays multiple roles in regulating cell functions, including fatty acid storage and glucose metabolism. The genes regulated by PPARγ stimulate lipid uptake and adipogenesis in adipocytes. In livers, PPARγ participated in the regulation of steatosis, which was induced by a high-fat diet in mice( Reference Inoue, Ohtake and Motomura 32 ). In Ashino et al.'s study( Reference Ashino, Saito and Souza 33 ), the expression of fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC) in the liver was significantly lower in adult offspring from dams that were fed a high-fat diet than that in the CD group, which corroborates our earlier finding that fatty acid synthesis was not stimulated by a maternal high-lipid diet in adult mice that were fed CD after weaning( Reference Yu, Miao and Gao 19 ). In the present study, global methylation in gene promoter sites might explain the low expression level of genes.
LXRα is another nuclear receptor that regulates the metabolism of several important lipids. The predominant function of LXRα is to regulate intracellular cholesterol balance by inducing the gene expression of cholesterol 7α-hydroxylase and ATP-binding cassette transporters( Reference Zhang, Chan and Cummins 34 ). In a rabbit model, the low expression of LXRα in the placenta was found in male offspring of dams that were fed high-lipid diets( Reference Tarrade, Rousseau-Ralliard and Aubrière 35 ). It has been reported that promoter sites were hypermethylated in mice offspring from dams that were fed protein-restricted diets during gestation( Reference van Straten, Bloks and Huijkman 36 ). In the present study, the expression of liver LXRα was inhibited by a maternal HLE diet, and hypermethylation at promoter sites might be involved.
In the present study, GO related to cell proliferation, protein phosphorylation, cell adhesion, apoptotic process and DNA replication were affected. Pathways related to cancer were significantly affected. The fetal developmental process is also a process of cell differentiation and proliferation( Reference Yu, Miao and Gao 19 ). The present study indicated the possibility that most cancer-related pathways were affected, and this hypothesis needs to be verified in the future.
In conclusion, the present study indicates that a maternal high-fat diet during gestation and lactation induced global DNA hypermethylation, including fatty acid and cholesterol metabolism-related genes.
Supplementary material
To view supplementary material for theis present article, please visit http://dx.doi.org/10.1017/S0007114515000252
Acknowledgements
The authors would like to acknowledge the technical assistance of the Genminix Company (Shanghai, China).
The present work was funded by grants from the Nature Science Foundation of China (81273070, 81102122 and 30901197).
The authors declared that there are no competing financial interests in relation to the present work. The authors state that there is no conflict of interest.
H.-L. Y. and R. X. conceived of and designed the experiments. S. D., L.-F. G., L. L., Y.-D. Xi. and W.-W. M. performed the experiments. S. D. and L.-H. Y. analysed the data. H.-L. Y. wrote the paper.








