
7 results
Staphylococcus epidermidis joint isolates: Whole-genome sequencing demonstrates evidence of hospital transmission and common antimicrobial resistance
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 45 / Issue 2 / February 2024
- Published online by Cambridge University Press:
- 15 December 2023, pp. 150-156
- Print publication:
- February 2024
-
- Article
- Export citation
Surveillance of healthcare-onset clinical cultures using whole-genome sequencing reveals hidden nosocomial transmission
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 3 / Issue S2 / June 2023
- Published online by Cambridge University Press:
- 29 September 2023, pp. s83-s84
-
- Article
-
- You have access
- Open access
- Export citation
-
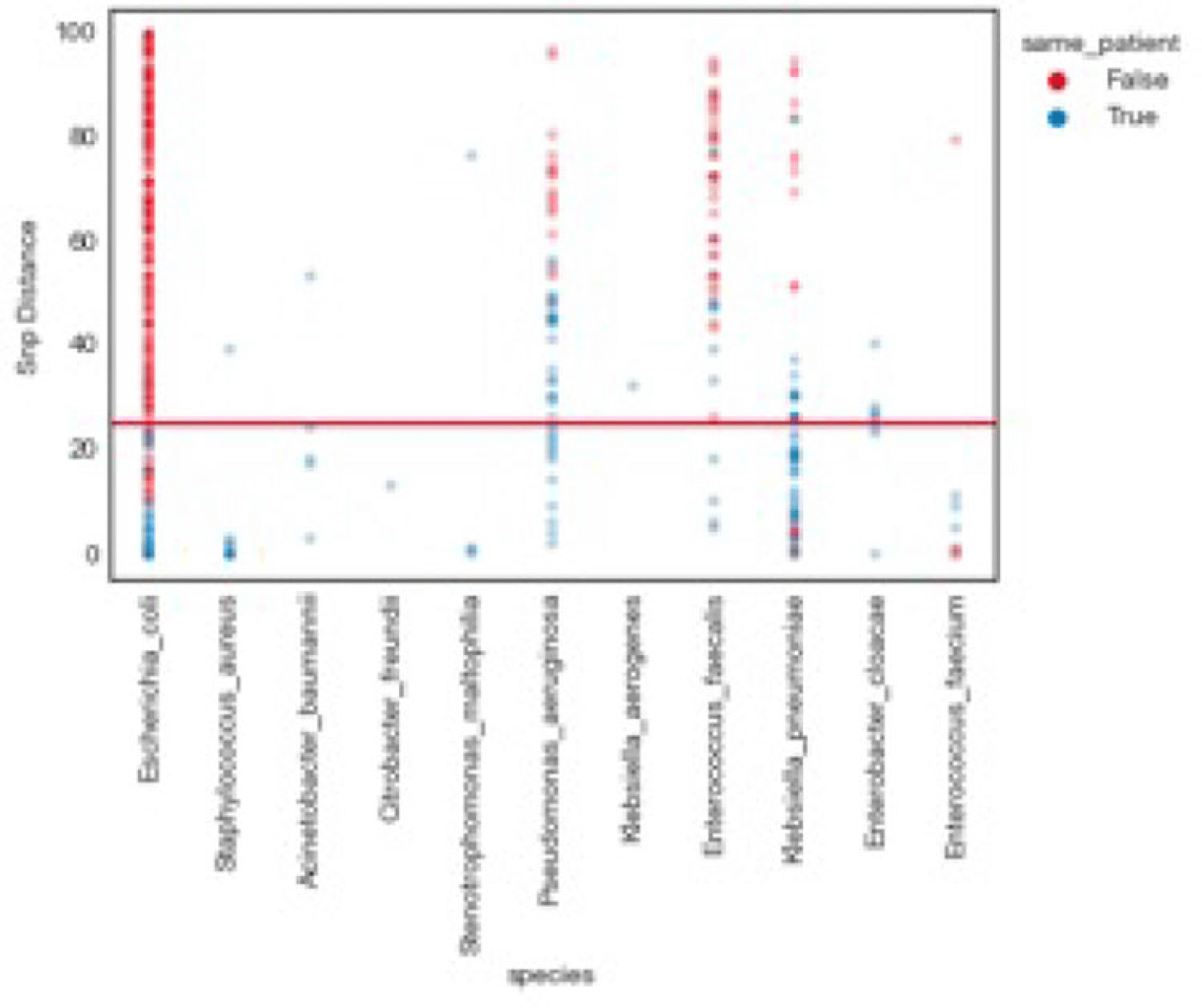
Background: Traditional hospital outbreak-detection methods are typically limited to select multidrug-resistant pathogens in a single unit, which can miss transmission of many medically important healthcare-transmissible pathogens. Whole-genome sequencing (WGS) enables comprehensive genomic resolution for accurate identification of clonal transmission. Previously, lack of scalability limited the use of WGS for hospital surveillance. Methods: We conducted prospective surveillance of select bacteria from all inpatient clinical cultures plus all bacteria from clinical cultures from ICUs and oncology units at the University of California Irvine (UCI) Clinical Microbiology Laboratory from September 2021 to February 2022. Due to pandemic stressors, this pilot test was a prelude to a real-time demonstration project. Its goal was to demonstrate the efficiency and scalability of the WGS platform when receiving samples monthly and analyzing results quarterly without the intent for real-time response. Bacterial isolates slated for discard were collected weekly and sent monthly to Day Zero Diagnostics for sequencing. In total, 1,036 samples from 926 patients were analyzed for genomic relatedness, a scalable and automated analysis pipeline already in use for rapid (days) characterization of genomic-relatedness in small and large sets of isolates. Mapping and SNP calling was performed against high-quality, best-match reference genomes. Sets of samples with pairwise distance of 2 persons with genomically related isolates and were denoted as “clusters.” Separately, we also investigated within-patient diversity by quantifying the genomic relatedness of isolates collected from individual patients. Results: Isolates represented 28 distinct species. We identified 10 Escherichia coli clusters (range, 2–4 patients; median, 2 patients), 2 Klebsiella pneumoniae clusters (range, 2–4 patients), and 1 Enterococcus faecium cluster (3 patients). All but 1 involved genomically matched isolates from multiple hospital locations. There were 4 Escherichia coli ST131 clusters spanning 4 months, including 1 with 4 patients across 3 different hospital locations. At a species level, there were distinct differences between the observed SNP distances between samples isolated from the same versus different patients (Fig. 1). All identified clusters had not been flagged by routine outbreak detection methods used by the UCI infection prevention program. Conclusions: Comprehensive WGS-based surveillance of hospital clinical isolates identified multiple potential transmission events between patients not in the same unit at the time cultures were taken. Combining WGS detection and real-time epidemiologic investigation may identify new avenues of transmission risk and could provide early warnings of clonal transmission to prevent larger outbreaks. High-volume surveillance of hospital isolates can also provide species- and context-specific clonality.
Financial support: This study was funded by Day Zero Diagnostics.
Disclosures: None

Detecting fecal microbiota transplantation–associated infection transmission using shotgun metagenomic sequencing and clonality analysis
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 3 / Issue S2 / June 2023
- Published online by Cambridge University Press:
- 29 September 2023, p. s86
-
- Article
-
- You have access
- Open access
- Export citation
Whole-genome sequencing to assess clonality in a series of prosthetic joint Staphylococcus epidermidis isolates – WITHDRAWAL
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 2 / Issue 1 / 2022
- Published online by Cambridge University Press:
- 04 October 2022, e163
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Whole-genome sequencing to assess clonality in a series of prosthetic joint Staphylococcus epidermidis isolates – WITHDRAWN
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 2 / Issue S1 / July 2022
- Published online by Cambridge University Press:
- 16 May 2022, pp. s54-s55
-
- Article
-
- You have access
- Open access
- Export citation
-
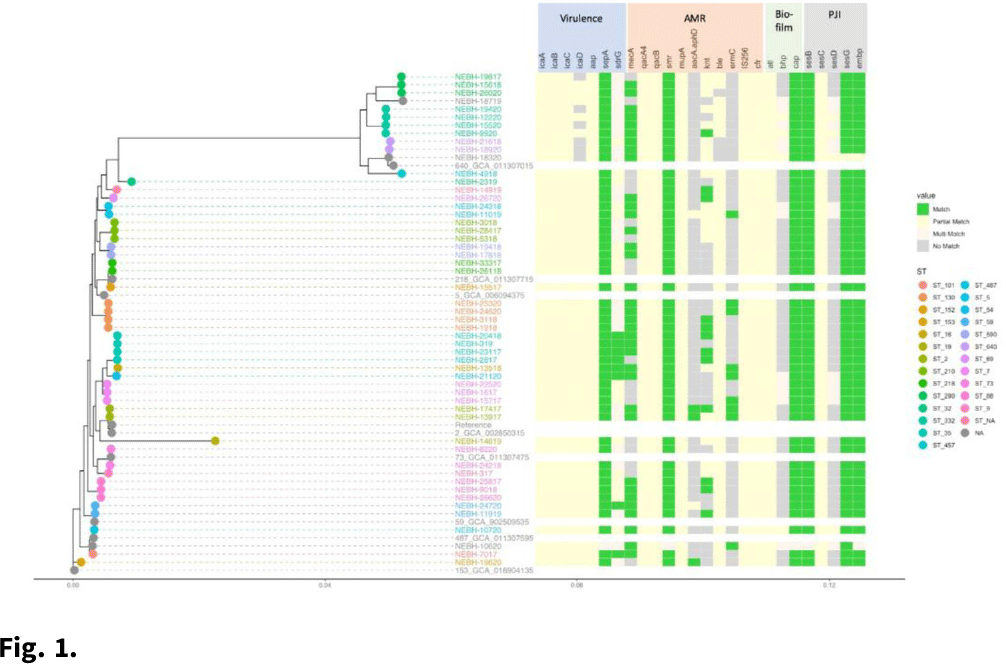
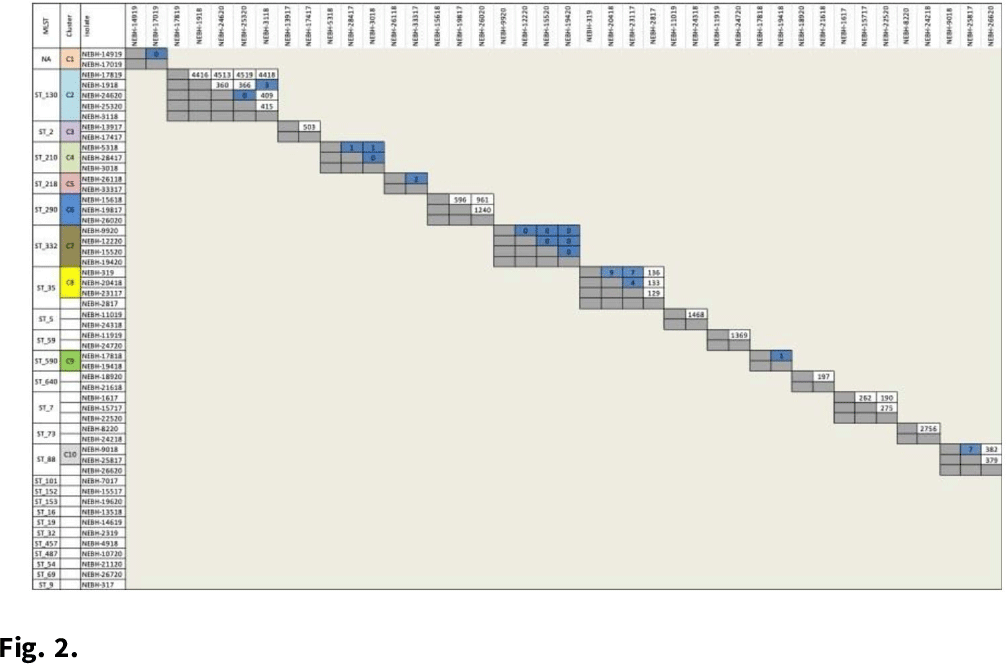
Background: Prosthetic joint infections (PJIs) are costly and cause increased morbidity and mortality for patients. Staphylococcus epidermidis is a common cause of both early postoperative and late-presenting PJIs. Although S. epidermidis is a normal part of the human skin microflora, its ability to form biofilm on implanted medical devices make it an important causative pathogen of PJIs. We investigated genetic, epidemiologic, and environmental factors contributing to S. epidermidis PJIs by performing whole-genome sequencing and clinical epidemiologic investigation of isolates collected from infected patients between 2017 and 2020. Methods: Patients with S. epidermidis isolated from a prosthetic joint that was placed at our orthopedic specialty hospital were identified using the microbiology laboratory records and electronic medical records. Whole-genome sequencing and single-nucleotide polymorphism (SNP)–based clonality analyses were performed using the epiXact service at Day Zero Diagnostics. These analyses included species identification, in silico MLST typing, phylogenomic analysis, as well as genotypic assessment of the prevalence of specific antibiotic resistance genes, virulence genes, and other relevant genes. For clonal isolates, additional reviews of surgical history and clinical data were performed. Results: In total, 62 S. epidermidis joint isolates were identified from 46 patients. Among these isolates, 52 were of sufficient purity to be used for genomic analysis (Fig. 1). A number of genes appeared in every isolate including sepA, smr, cap, sesB, sesG, and embp. Also, 6 S. epidermidis samples had a discrepancy between phenotypic resistance to oxacillin and the presence of the mecA resistance gene. We also identified 6 distinct clusters of isolates, all of which had SNP distances <10 base pairs (Fig. 2). Each cluster consisted of 2–4 patients. Cluster isolates accounted for 29.8% of all S. epidermidis prosthetic joint isolates. Most clonal isolates occurred in patients who were heavily exposed to different healthcare settings. Further epidemiologic investigation showed that some of these clonal isolates had ties to aspirations or procedures, whereas no clear connection could be determined for others. Conclusions:S. epidermidis isolated from clinical prosthetic joint samples contains a high degree of genetic resistance, including a mismatch between presence of mecA and phenotypic oxacillin resistance and genetic propensity for chlorhexidine resistance. Mupirocin resistance was not observed. Of all isolates, 29.8% belonged to multiple clusters, confirming hospital spread of this commensal organism in some cases.
Funding: None
Disclosures: None


Democratizing Sequencing for Infection Control: A Scalable, Automated Pipeline for WGS Analysis for Outbreak Detection
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue S1 / October 2020
- Published online by Cambridge University Press:
- 02 November 2020, pp. s442-s443
- Print publication:
- October 2020
-
- Article
-
- You have access
- Export citation
Community-acquired in name only: A cluster of carbapenem-resistant Acinetobacter baumannii in a burn intensive care unit and beyond
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue 5 / May 2020
- Published online by Cambridge University Press:
- 28 February 2020, pp. 531-538
- Print publication:
- May 2020
-
- Article
- Export citation
