

At present, the treatment options for Alzheimer's disease are largely symptomatic giving rise to temporary remissions in cognitive decline or the amelioration of troublesome behavioural and psychological symptoms. However, as more is becoming known about the molecular biology of Alzheimer's disease, there is a clear movement to treatments directed towards disease modification. Disease modification is now a realistic option and its importance is highlighted by the fact that a delay in the progression of the disease by just five years would halve the disease prevalence. Ultimately, however, drug therapy based on the molecular biology of Alzheimer's disease will be preventive. These treatment strategies are still in their infancy, but our understanding of the processes requiring therapeutic manipulation is improving at a dramatic rate.
Prevention
Direct support for a genetic component to Alzheimer's disease comes from the recognition of a small number of patients who develop the disease largely before the age of 65 years in a clear autosomal dominant pattern. To date, mutations in three genes (amyloid precursor protein (APP), presenilin 1 (PS1) and presenilin 2 (PS2)) have been described which lead to this early form of Alzheimer's disease. Pathogenic APP, PS1 and PS2 mutations have been shown to alter the proteolytic processing of amyloid precursor protein giving rise to an increased production of the β-42 form of amyloid protein, which forms the core of neuritic plaques and which has been shown to be neurotoxic to cells in culture (Reference Lambert, Barlow and ChromyLambert et al, 1998). In addition, presenilin mutations may increase neuronal vulnerability by altering the stability or intracellular trafficking of β-catenin, a protein known to be involved in neuronal apoptosis (programmed cell death) (Reference Nishimura, Yu and LevyesqueNishimura et al, 1999).
In Alzheimer's disease developing after the age of 65, no clear autosomal dominant patterns have been established. However, a wide number of different candidate genes have been proposed, variations in which have been associated with an increased risk of developing Alzheimer's disease. Of these candidates, the most substantially corroborated genetic risk factor remains that of the presence of the common polymorphism apolipoprotein E ϵ4 (ApoE ϵ4). The role of ApoE ϵ4 in the molecular pathogenesis of Alzheimer's disease has yet to be clearly established, but the inheritance of one or two ApoE ϵ4 alleles may result in enhanced aggregation and/or decreased clearance of β-amyloid (Reference Schmechel, Saunders and StrittmatterSchmechel et al, 1993).
Direct genetic manipulation using therapeutic gene transfer in Alzheimer's disease is still largely theoretical, but the technical prerequisites are being developed at an astonishing rate (Reference LeschLesch, 1999). Two basic procedures, the ex vivo and in vivo approaches, have been developed. In the ex vivo procedure, the transgene is introduced into the cells by a viral vector in vitro, which is then grafted into the organism. This approach may enable the replacement of neurotransmitters or neurotrophic growth factor, by grafting in modified primary neuronal, stem or progenitor cells. In the in vivo approach, the gene is delivered into the organism by a viral vector for in situ transduction of the target brain cell. This latter approach has a more immediate application in understanding the role of genetic risk factors in animal models, rather than a direct role in the treatment of Alzheimer's disease, where in the majority of cases the aetiology is multifactorial and polygenic.
Box 1. Strategies for prevention
Direct genetic manipulation
Inhibition of β-amyloid production
Inhibition of β-amyloid aggregation
Other preventive strategies are to aim treatments at the early stages of the biological cascade in Alzheimer's disease. These could include the use of compounds that inhibit β-amyloid production by reducing the activity of enzymes, including β- or γ-secretase, which cleave β-amyloid from APP, or by the use of inhibitors of β-amyloid aggregation or fibrillisation. Another possible approach has been highlighted in a recent report (Reference Schenk, Barbour and DunnSchenk et al, 1999), which showed that in a mouse model of Alzheimer's disease immunisation with β-amyloid inhibits the formation of β-amyloid and its associated dystrophic neurites in mouse brain.
Whichever approach turns out to be successful in patients with Alzheimer's disease, it is clear that other problems will need to be overcome before a preventive strategy can be put in place. These include the availability of clinical or biological parameters for early diagnosis, screening procedures, the monitoring of therapeutic response and the all-important question of cost.
Arresting progression
Based largely on studies of patients with Down's syndrome and transgenic mice that express mutant APP and/or PS1, it is hypothesised that β-42 accumulation and the consequent neuritic plaque formation is associated with microglia activation, reactive astrocytosis and a widespread inflammatory response. It is thought that this inflammatory response, coupled with the direct toxicity arising from oligomeric and fibrillar β-amyloid, produces the structural and consequent neurochemical changes seen in Alzheimer's disease. In addition, it is thought that the effects of this inflammatory and neurotoxic process are then further compounded by the excess generation of free radicals and peroxidative injury, which causes further damage to neurons (Reference Harris, Hensley and ButterfieldHarris et al, 1995).
It follows that drugs that interfere with one or a number of these processes could lead to arrest or delay in the progression of the disease. Three approaches can be envisaged.
-
(a) Use of anti-inflammatory drugs that could interfere with the inflammatory responses in the brain.
-
(b) Use of antioxidants and free radical scavengers that could protect neurons from the downstream effects of the accumulation of β-amyloid.
-
(c) Use of neuroprotective factors that could repair cell damage or restore damaged synapses.
Anti-inflammatory agents
An early study suggesting an inverse relationship between the presence of rheumatoid arthritis and Alzheimer's disease led to speculation that this association was owing to the higher use of non-steroidal anti-inflammatory drugs (NSAIDs) in the arthritic group (Reference McGeer, Schulzer and McGeerMcGeer et al, 1996). Since then, more than 20 epidemiological studies have indicated that patients taking anti-inflammatories, or those suffering from conditions where such drugs are routinely used, have a substantially reduced risk of developing Alzheimer's disease. However, the risks of non-selective NSAID use, including gastric irritation, peptic ulceration and nephrotoxicity, have led to interest, and clinical trials, in other anti-inflammatory agents. These agents include prednisolone, hyroxychloroquine, colchicine and selective cyclooxygenase-2 (COX-2) inhibitors. Primary analysis of data from a study using moderate-dose prednisolone appears to show no evidence of a reduction in cognitive decline, and use of higher doses of the drug was limited following the problematic side-effects. Of the other drugs under trial, particular interest is being paid to the use of COX-2 inhibitors since, as well as potentially offering a reduced risk of toxicity, there is some evidence to suggest that the neuronal expression of COX-2 is up-regulated in the brain of people with Alzheimer's disease and that COX-2 activity increases the susceptibility of neurons to amyloid toxicity.
Antioxidants
Different sources of oxidative stress in Alzheimer's disease suggest several pharmacological approaches to influence disease progression. Two major types of therapeutic agent can be described according to their pharmacological point of attack.
-
(a) Radical scavengers – agents directly interacting with free radicals. These include gingko biloba, vitamins A, C and E, and oestrogen.
-
(b) Antioxidants, which are able to prevent or decrease the production of free radicals. These include selegiline and tenilsetam.
In vitro studies have demonstrated the efficacy of both types of therapeutic agents by preventing or delaying oxidative neuronal damage. Some clinical data also exist regarding the use of gingko biloba (Reference Le Bars, Katz and BermanLe Bars et al, 1997), selegiline and vitamin E (Reference Sano, Emesto and ThomasSano et al, 1997). In the latter study, vitamin E and selegiline demonstrated marginal superiority to placebo in slowing functional deterioration in patients with moderately advanced Alzheimer's disease. Thus, in general, efficacy studies using these compounds seem to indicate a possible future strategy in the therapy of Alzheimer's disease. However, it is too early to draw definite conclusions about these compounds since it is clear that these candidates have a number of properties and do not act specifically as radical scavengers or antioxidants.
Neuroprotective factors
The use of neurotrophins, which are needed to maintain cholinergic pathways in the brain, is an attractive proposition, but failure to cross the blood–brain barrier causes immense practical difficulties. Modification of these compounds or the use of smaller molecules which mimic their action, such as oestrogen, is however more promising. Oestrogen has a number of beneficial functions for patients with Alzheimer's disease, including antioxidant, anti-amyloid and possible anti-inflammatory properties. However, there is also some evidence to suggest that its effects include the promotion of the growth and survival of cholinergic neurones. Thirteen retrospective studies have looked at patients previously receiving oestrogen replacement therapy and their subsequent risk of developing Alzheimer's disease. Of these studies, nine showed a decreased risk but two failed to highlight any difference and two showed an increased risk. Of four treatment studies, with a total of 58 women, only one study was a randomised placebo-controlled trial. In this study of 12 cognitive tests, six showed improvement. In the retrospective studies, confounders such as increased education and better health in women taking oestrogen supplements have not been adequately controlled for. In the treatment studies, in addition to the small numbers involved, it is clear that there is considerable heterogeneity between studies with a wide variety in the type and duration of oestrogen replacement. Thus, in addition to the paucity of studies, it is clear that there are a number of limitations to the published studies which make it impossible to advocate the use of oestrogen therapy as yet (see Reference Behl and HolsboerBehl & Holsboer, 1999).
Box 2. Strategies for arresting progression
Use of anti-inflammatory drugs
Use of antioxidants/free radical scavengers
Use of neuroprotective factors
Symptomatic treatment
Following neuronal cell damage and loss by apoptosis come inevitable changes in brain neurochemistry. Symptomatic treatments are largely based on our understanding of these neurochemical changes. Many neurotransmitter systems are affected in Alzheimer's disease, but prominent losses are seen in the neurotransmitters acetylcholine, glutamate and serotonin. Therapeutic optimism surrounding dopamine replacement in parkinsonism led to early hypotheses that replacement of such neurotransmitter losses, particularly acetylcholine, might have a role to play in the treatment of Alzheimer's disease, and this approach is still the major thrust of pharmacological treatments for Alzheimer's disease.
More recently, the approach has gained further support from the finding that such a strategy might also have an influence on the production of β-amyloid plaques by stimulation of the secretory pathway.
Amyloid precursor protein can be processed by several alternative proteolytic mechanisms. In the secretory pathway, APP is cleaved by α-secretase within the β-amyloid domain, leading to the production of a soluble fragment (APPs), which is secreted into the extracellular space. Amyloid precursor protein molecules that escape α-secretase cleavage can be re-internalised from the cell surface and targeted to an acidic endosomal–lysosomal compartment, where they are processed by different set of enzymes that cleave both at the N-terminus of β-amyloid (β-secretase) and at its C-terminus (γ-secretase). These cleavage events produce intact 40- to 42-residue β-amyloid molecules that can aggregate and form amyloid plaques. Amyloid precursor protein processing is under the control of G-protein-coupled neurotransmitter receptors that increase α-secretase processing and inhibit β-secretase processing. As a result, stimulated cells secrete more APPs and less β-amyloid compared with unstimulated cells. A number of receptors have been shown to stimulate the α-secretase pathway including the M1 and M3 acetylcholine receptor subtypes, the serotonin receptor subtypes 5-HT2a and 5-HT2c and the metabotropic glutamate mGluR1a-receptor. Thus, drugs which directly or indirectly stimulate these receptors could have short-term as well as long-term benefits by slowing down the production of neuritic plaques.
Acetylcholine
Of the neurotransmitter losses, the first to be recognised was that of acetylcholine. The reduction in acetylcholine (of around 50%) is associated with presynaptic neuronal losses of a similar order, suggesting that neurochemical deficits are largely a reflection of neuronal cell loss. To date, therapeutic strategies in Alzheimer's disease aimed at increasing available acetylcholine include the use of acetylcholine precursors, acetylcholine releasers, acetylcholine agonists and acetylcholinesterase inhibitors.
Acetylcholine precursors
One of the first strategies was an attempt to increase acetylcholine production using precursor loading. However, repeated studies have failed to produce positive results – although a recent Cochrane review (Reference Fioravanti and YanagiFioravanti & Yanagi, 1998) of mixed dementias suggests some evidence of positive effect in the short term for choline.
Acetylcholine releasers
A number of drugs have been regarded as acetylcholine releasers including a few pyridine derivatives, which are predominantly potassium channel-blocking agents. One drug, linopirdine, showed no advantage over placebo and was later found to have a releasing effect on acetylcholine only in the presence of high potassium concentrations, suggesting limited use in vivo. Another, besipirdine, a selective M-channel blocker, was found not to be an effective acetylcholine releaser and trials have been unremarkable.
Box 3. Symptomatic treatments
Based on replacement of the neurotransmitter losses seen in Alzheimer's disease
Based on the manipulation of three major neurotransmitter systems: acetylcholine, glutamate and serotonin
Manipulation of the acetylcholine system, particularly by the use of cholinesterase inhibitors, has proven to be a successful strategy in delaying cognitive deterioration in patients with Alzheimer's disease
Manipulation of the serotinergic system by the use of atypical antipsychotics has proven to be a successful strategy in the treatment of some non-cognitive symptoms found in Alzheimer's disease
Acetylcholine receptor agonists
There are a number of orally active selective muscarinic agonists that have reached clinical trials, but despite therapeutic optimism, results have been disappointing. These drugs include xanomeline, sabcomeline, milameline and talsaclidine.
Xanomeline is an M1 and M4 selective agonist and the first to show a significant though modest treatment effect on cognition as measured by the cognitive sub-scale of the Alzheimer's Disease Assessment Scale (ADAS–cog). Interestingly, this drug also showed an improvement on a wide variety of non-cognitive symptoms including hallucinations, agitation, vocal outbursts, delusions, suspiciousness and mood swings (Reference Bodick, Offen and LeveyBodick et al, 1997). Unfortunately, because of unacceptable side-effects (nausea, vomiting and fainting) the oral preparation was dropped and phase II trials using transdermal patches are underway in the hope of eliminating the troublesome metabolite produced in the gut. Sabcomeline is a partial M1 agonist and showed some cognitive efficacy in a controlled trial of 364 patients over 14 weeks (Reference Kumar and OrgogozoKumar & Orgogozo, 1996). Milameline, an M1/M2-receptor agonist, has been disappointing and trials abandoned. Talsaclidine is a functionally selective M1-receptor agonist currently in phase II trials.
More recently, attention has been directed at the possible role of nicotinic receptor modulation for the treatment of Alzheimer's disease. Nicotinic agonists, often thought too toxic, are arousing interest particularly as galantamine, one of the cholinesterase inhibitors that has shown efficacy in clinical trials, is thought to stimulate nicotinic receptors through allosteric modulation. Other nicotinic modulators currently in preclinical development include GTS-21, a nicotinic agonist that facilitates learning and memory in animals.
Cholinesterase inhibitors
This group of drugs has shown consistent efficacy and to date they are the only cholinomimetics to have gained regulatory approval for treating Alzheimer's disease. At present, four compounds have gained licences in some European countries: tacrine, donepezil, rivastigmine and galantamine.
Tacrine (tetrahydroaminoacridine or THA) is a reversible inhibitor of both acetylcholinesterase (AchE) and butyrylcholinesterase (BuChE), which requires four-times-daily dosing. A 30-week trial which tested the compound at higher doses than previous trials found significant improvements over placebo in the ADAS-cog, Global Assessment Scale and Mini-Mental State Examination (Reference Knapp, Knopman and SolomanKnapp et al, 1994). However, tolerability and side-effects (principally liver transaminitis) limited its usefulness.
The short action and poor tolerability of physostigmine have prevented it from becoming an acceptable treatment and as yet it is not licenced. The heptyl analogue of physostigmine, heptastigmine, has been found to be more stable, longer-lasting, if less potent, and significantly less toxic than the former. In phase III studies, it has shown typical AChE efficacy in terms of improvement in the ADAS–cog. Clinical development however, has been slow, and one company withdrew after a neutropenia developed in two patients (Reference Enz and GauthierEnz, 1998).
The drug with the greatest impact so far on the treatment of Alzheimer's disease has been donepezil. Donepezil is 10 times less potent than physostigmine, but this has not diminished its efficacy in terms of cognitive function, global assessment and activities of daily living. The three main placebo-controlled studies (Reference Rogers and FriedhoffRogers & Friedhoff, 1996, Reference Rogers and Friedhoff1998; Reference Rogers, Doody and MohsRogers et al, 1998) all show an improvement in cognition over baseline at the end of six months of around one point on the ADAS–cog with an end-point effect size of just under three points. Long-term follow-up studies seem to suggest (although without a placebo group it is only a suggestion) that the cognitive advantage over placebo is maintained for up to two years (Reference Rogers and FriedhoffRogers & Friedhoff, 1998). There is also now clear evidence emerging from clinical practice that changes in behavioural or non-cognitive symptoms are also a feature of response to donepezil (Reference Burns, Rossor and HeckerBurns et al, 1999).
Rivastigmine is licensed in the UK for the symptomatic treatment of Alzheimer's disease. A carbamate derivative of physostigmine, its effects are similar to those of eptastigmine. It does not inactivate acetylcholine by the usual microsomal activity but by attaching a carbamyl residue, which means that although it has a half-life of two hours, cholinesterase inhibition in the brain is thought to last for up to 10 hours. It is, therefore, classed as a pseudo-irreversible inhibitor and is highly specific for AChE. The phase III clinical trials of rivastigmine have now included some 3000 patients and these studies suggest an effect size of 3.8 ADAS points (Reference Corey Bloom, Anand and VeachCorey Bloom et al, 1998), though with a general deterioration in all groups. The fact that 20% of the high-dose group had improved by seven points or greater on the ADAS-cog suggests that the effect size was a genuine reflection of efficacy in a worsening group of patients. The main drawbacks of rivastigmine are its short half-life, the consequent twice-daily dosing, and the necessity for slow titration to minimise the cholinergic side-effects. The trial data suggest these are not severe but include nausea, vomiting and anorexia. Rivastigmine has also been linked to improvements in non-cognitive symptoms in a small open-label study of patients suffering from dementia with Lewy bodies. They found that the Neuropsychiatric Inventory sub-scale scores for apathy, delusions, hallucinations and agitation fell by 82% in six of the 11 patients in the study (further details available from the author upon request).
Metrifonate is a simple organophosphorous compound which is slowly hydrolysed to dichlorvos in the body – a potent irreversible cholinesterase inhibitor. This slow metabolism and permanent inactivation of the enzyme means that once-daily dosing is sufficient. Pooled data in US studies showed an effect size of 3.8 ADAS–cog points with an improvement over baseline of 1.8 points, the latter comparable with the best in its class (further details availavle from the author upon request). The European Metrifonate in Alheimer's Trial (MALT) study showed similar efficacy on cognition, significant changes in behaviour and function and, for the first time in a placebo-controlled study, significant improvements in carer burden. Unfortunately, axial and proximal muscle weakness has been noted in a number of patients in the USA and the trial programme has been halted while this is investigated. It has been severe enough to cause respiratory failure in a few patients who had been on the drug for a long time.
Galantamine is a natural alkaloid and shares with this group the potential to allosterically potentiate submaximal nicotinic responses induced by acetylcholine and competitive agonists. It may well be that this nicotinic effect is the reason why the AChE inhibitors (AChEIs) are more effective than other cholinomimetics. Allosteric potentiating ligands like galantamine could, therefore, enhance nicotinic transmission under conditions of reduced secretion or increased degradation of acetylcholine, or where the acetylcholine sensitivity of nicotinic acetylcholine receptors is reduced. This may well mean that they will have a preventive and corrective action on impaired nicotinic transmission and may have more than a symptomatic effect on Alzheimer's disease itself. Studies so far have certainly shown evidence of cognitive improvement – the two most recent phase III studies in USA and the international phase III trial all showing remarkably consistent results with effect sizes of 3.7 and 4.1 points on the ADAS–cog, respectively, with improvement of 1.7 points from baseline at six months (further details available from the author upon request). Data from the US open-label continuation study indicate maintenance of this benefit with no decrement from baseline a year later.
All studies also showed improvements in the global assessment and functional ability. What is particularly interesting in many of these trials is the proportion of patients who show quite marked improvements in cognition. In a phase II study of galantamine, 10% of patients on the 30 mg dose showed an improvement of 15 points or greater on the ADAS–cog after 12 weeks (further details available from the author upon request). Galantamine, like all AchEIs, does have tolerability problems unless titrated slowly and, as expected, these are predominantly gastrointestinal. It has, however, over 90% bio-availability and so can be given with food. It is a competitive, reversible and specific inhibitor of acetylcholine, with a half-life of about eight hours, and needs to be given twice-daily. It seems likely that it may be licensed in the UK and may show benefits over existing drugs, but long-term effects remain to be seen.
Glutamate
Beta-amyloid has been shown to alter calcium homoeostasis in human cortical neurons. This rise in internal calcium has been linked to a number of biochemical processes, including the activation of a series of enzymes (protein kinase C, calcium calmodulin-dependent protein kinase II, phospholipases, proteases, phosphataes, nitric oxide synthase and orthinine decarboxylase) that lead to neuronal cell death by protein breakdown, free radical formation and lipid peroxidation. Excessive stimulation of the ionotropic glutamate receptors N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA) has also been shown to lead to an excessive influx of calcium as well as the release of intracellular calcium stores.
A number of therapeutic interventions are possible which may offset this excitotoxic cascade. These include a reduction in the synthesis and release of glutamate, an increase in glutamate uptake, decrease in glutamate receptor antagonists and inactivation of the secondary intracellular events. However, it is also clear that a complete blockade of glutamatergic neurotransmission has adverse effects on the higher activities of glutamate in the cerebral cortex, which include memory and learning. Drugs proposed that might fulfil these dual roles include partial agonists of the strychnine-insensitive glycine site of NMDA, positive modulators of AMPA receptors and non-competitive NMDA receptor antagonists.
Recent preclinical studies show promising results with ampakines, such as 1,3-benzodiaoxol-5-ylcarbonyl piperidine (BDP), which enhance excitatory post-synaptic potentials and AMPA-gated currents and have been shown to improve memory and reduce age-related cognitive deficits in animals and humans (Reference Lynch, Kessler and RogersLynch et al, 1996). Memantine, a non-competitive NMDA receptor antagonist, has been used clinically in Europe for some time. Early studies suggested that patients with dementia treated with memantine in doses of 10–20 mg/day showed mild improvements in a wide variety of parameters including memory, mood, behaviour and activities of daily living. More recently, significant improvements in moderate to severe dementia, as assessed by global, behavioural and activities of daily living end-points, have been reported (Reference Winblad and PoritisWinblad & Poritis, 1998). Most of these studies have been carried out in patients with mixed dementias, thus it appears unclear whether there is any disease specificity. In addition, patients in these studies had dementia of mild to moderate severity and it may be, given the potential role of glutamate in initiating neurodegeneration, that early treatment may have also had a neuroprotective effect.
Serotonin
Although reductions in the acetylcholine and glutamatergic systems have been largely associated with alterations in cognition in Alzheimer's disease, losses in the serotonergic system and to a lesser extent the noradrenergic and dopaminergic systems have been associated with a wide variety of behavioural and psychological disturbance seen in Alzheimer's disease.
Recently, attention has turned to the adverse extrapyramidal or anticholinergic side-effects that conventional neuroleptics have in the treatment of behavioural disturbance and psychosis in Alzheimer's disease. This has led to an increased interest in the potential use of atypical neuroleptics, including clozapine, olanzapine, quetiapine and risperidone. These drugs have a greater affinity for 5-HT2 receptors than D2 receptors in the brain, which is thought to account for the lower incidence of extrapyramidal side-effects. Both clozapine and olanzapine have high affinity for antagonism at muscarinic M1 receptors, where quetiapine has little and ripseridone no affinity.
Relatively few studies have looked at the effects of clozapine in patients with Alzheimer's disease and, in general, although it appears effective for psychotic symptoms, its use is limited because of side-effects including agranulocytosis and predictable anticholinergic actions.
To date, two studies have looked at the effects of olanzapine in patients with Alzheimer's disease (Reference Satterlee, Reams and BurnsSatterlee et al, 1995; further details available from the author upon request). The first study using small doses (1–8 mg/day) failed to find any increased efficacy above placebo. The second double-blind placebo-controlled study found that both 5 and 10 mg/day were significantly superior to placebo in reducing psychotic and behavioural symptoms and, surprisingly, given the potential anticholinergic effects, there was no evidence of cognitive decline or peripheral anticholinergic side-effects (Reference Arvanitis and RakArvanitis & Rak, 1997).
Only one non-blind study (Reference Arvanitis and RakArvanitis & Rak, 1997) has so far looked at the effects of quetiapine in patients with mixed dementia and organic psychoses. This study showed improvements in the Brief Psychiatric Rating Scale in patients and appeared well-tolerated, although clearly large double-blind trials are needed to properly evaluate its efficacy and tolerability in Alzheimer's disease.
To date, the largest number of studies of atypical antipsychotics have looked at the effects of risperidone on behavioural and psychological disturbances in dementia. These include case reports, non-blind trials, structured trials, retrospective chart reviews as well as two large double-blind placebo-controlled studies. In the double-blind placebo-controlled studies (Reference Katz, Jeste and MintzerKatz et al, 1999; further details available from the author upon request), risperidone was shown to produce significantly greater reductions in the overall severity of behavioural symptoms and of psychosis and aggressivity in patients with Alzheimer's disease. Optimal dosage appears to be between 1 and 2 mg/day, since doses of 2 mg/day had an increased frequency of extrapyramidal side-effects but 1 mg/day was indistinguishable from placebo. Risperidone was not shown to be superior to haloperidol in the treatment of aggression in Alzheimer's disease but it does appear to have significantly less extrapyramidal side-effects. This finding has been emphasised in two longitudinal studies (Reference Jeste, Lacro and BaileyJeste et al, 1999; further details available from the author upon request). Thus, a one-year extension of the initial double-blind placebo-controlled study showed a lower incidence of tardive dyskinesia than would have been expected from conventional neuroleptics. In addition, a nine-month prospective study comparing haloperidol with risperidone showed that low-dosage risperidone was significantly less likely to produce tardive dyskinesia than low-dosage haloperidol.
Conclusions
There are considerable reasons for optimism in the treatment of Alzheimer's disease. Treatment strategies are moving rapidly from the symptomatic towards disease modification and indeed some of the current symptomatic treatments already have this potential. In the near future, it is likely that a combination of therapies aimed at both disease modification and symptomatic treatment will be in place. Ultimately, treatment will be preventive but there are a number of biological and clinical issues to be addressed before this becomes reality.
Multiple choice questions
-
1. The following drugs are all cholinesterase inhibitors:
-
a memantine
-
b donepezil
-
c galantamine
-
d rivastigmine
-
e risperidone.
-
-
2. Preventive strategies in treatment of Alzheimer's disease include:
-
a therapeutic gene transfer
-
b the use of cholinesterase inhibitors
-
c the use of atypical antipsychotics
-
d the reduction of β-secretase
-
e the reduction of activity of β-secretase.
-
-
3. A possible delay in progression of Alzheimer's disease might arise out of:
-
a the use of anti-inflamatory agents
-
b radical scavengers
-
c antoxidants
-
d cholinesterase inhibitors
-
e atypical antipsychotics.
-
-
4. Regarding the use of oestrogen in the treatment of Alzhiemer's disease:
-
a oestrogen may act as a radical scavenger
-
b oestrogen may have anti-amyloid properties
-
c oestrogen may have anti-inflamatory properties
-
d oestrogen may promote the survival of cholinergic neurons
-
e there is sufficient evidence to suggest the use of oestrogen therapy in a clinical setting.
-
-
4. Five drugs are atypical antipsychotics, with a potential role to play in the treatment of behavioural disturbance and psychosis in Alzheimer's disease:
-
a clozapine
-
b risperidone
-
c olanzapine
-
d galantamine
-
e melameline.
-
MCQ answers
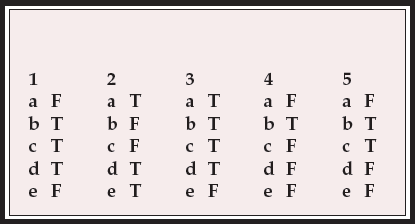
| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | F | a | T | a | T | a | F | a | F |
| b | T | b | F | b | T | b | T | b | T |
| c | T | c | F | c | T | c | F | c | T |
| d | T | d | T | d | T | d | F | d | F |
| e | F | e | T | e | F | e | F | e | F |

eLetters
No eLetters have been published for this article.