


- HDLC
HDL-cholesterol
- LDLC
LDL-cholesterol
The impact of single nucleotide polymorphisms on risk of chronic diseases such as CVD, and the ability of dietary factors to manipulate genotype–phenotype associations, is being increasingly recognised. Undoubtedly, the most-widely-studied gene variant in relation to CVD is the apoE ε (ε2, ε3, ε4) genotype. Since its discovery in 1973 the central role of the apoE protein in lipoprotein metabolism has been comprehensively investigated and reported. The 40–50% higher risk of CVD in apoE4 carriers (Song et al. Reference Song, Stampfer and Liu2004) has been traditionally attributed to moderately higher circulating cholesterol and TAG levels. However, it is becoming increasingly recognised that an effect on lipoprotein metabolism alone cannot explain the disease differential and that the impact of an apoE4 genotype is largely lipoprotein independent. Roles of macrophage-derived apoE protein on vascular health and atherogenesis are being identified, with apoE thought to impact on oxidative status and in an autocrine and paracrine manner affect macrophage, vascular smooth muscle cell, endothelial cell and platelet function. An impact of genotype on these localised functions of apoE could in part explain the impact of genotype on CVD pathology, as will be discussed.
Additionally, apoE genotype has been shown to affect the responsiveness to the total fat content and fatty acid composition of the diet. Manipulation of dietary fat content may serve as a means of reducing the increased CVD burden associated with an apoE4 genotype.
ApoE structure and tissue sources
ApoE was first described as a component of VLDL in the circulation (Shore & Shore, Reference Shore and Shore1973). The full amino acid sequence was elucidated in 1982, with the mature 299 amino acid 34 kDa acid protein resulting from the proteolytic cleavage of the 317 amino acid product of the apoE gene (Rall et al. Reference Rall, Weisgraber and Mahley1982). ApoE is found in the circulation associated with chylomicrons, VLDL and HDL at a typical concentration of 20–60 mg/l (Bhatnagar & Durrington, Reference Bhatnagar and Durrington1993).
The protein assumes a typical apo form with two structural domains (Fig. 1; from Hatters et al. Reference Hatters, Peters-Libeu and Weisgraber2006). The amino terminal (22 kDa) comprises residues 1–191 and ‘houses’ the lysine- and arginine-rich receptor-binding region contained between amino acids 136 and 150 (Innerarity et al. Reference Innerarity, Friedlander, Rall, Weisgraber and Mahley1983). The carboxyl terminal (10 kDa) consists of residues 225–299 and contains the major lipid-binding determinants that anchor apoE to the lipoprotein (Wetterau et al. Reference Wetterau, Aggerbeck, Rall and Weisgraber1988). These domains are separated by a protease-sensitive hinge region (Wetterau et al. Reference Wetterau, Aggerbeck, Rall and Weisgraber1988). Despite the independent folding of the two domains, there are recognised domain interactions (Dong & Weisgraber, Reference Dong and Weisgraber1996).

Fig. 1. Key structural elements of apo E (reprinted from Hatters et al. Reference Hatters, Peters-Libeu and Weisgraber2006, with permission from Elsevier). (a) The amino-terminal domain consists of a four-helix bundle that contains the LDL receptor-binding region of the protein contained between amino acids 136–150 in helix 4. Contained within the ‘hinge region’, amino acid 172 is thought to be essential for receptor binding. The carboxyl-terminal contains the lipoprotein-binding region. (b) The model demonstrates the impact of the replacement of Cys with Arg on position 112 in the protein. This replacement facilitates the interaction between Arg 61 and Glu 255, which mediates closer contact between the amino-terminal and carboxyl-terminal domains.
The structure of the carboxyl-terminal domain is unknown but is predicted to be mostly α-helical (Nolte & Atkinson, Reference Nolte and Atkinson1992), whilst the three-dimensional structure of the amino-terminal domain in lipid-free solution has been determined by X-ray crystallographic studies to be an elongated globular four-helix bundle. Helix 1 pairs with helix 2, and helix 3 with helix 4, arranged in an anti-parallel mode, with the hydrophobic faces oriented towards the interior of the bundle (Wilson et al. Reference Wilson, Wardell, Weisgraber, Mahley and Agard1991). ApoE genotype impacts on the three-dimensional orientation of the apoE regions and amino-terminal–carboxyl-terminal interactions, which affect receptor binding and lipoprotein apoE distribution, as will be discussed (see pp. 185–186). For more detailed information on apoE structure and structure–function relationships, see Hatters et al. (Reference Hatters, Peters-Libeu and Weisgraber2006).
ApoE is synthesised mainly in the liver, with hepatocytes being the main producers. It has been estimated that between 20 and 40% of the total apoE protein is produced by extrahepatic tissues, with the brain and the monocyte-derived macrophages expressing relatively high amounts (Basu et al. Reference Basu, Ho, Brown, Bilheimer, Anderson and Goldstein1982; Kayden et al. Reference Kayden, Maschio and Traber1985; Newman et al. Reference Newman, Dawson, Rudel and Williams1985; Wang-Iverson et al. Reference Wang-Iverson, Gibson and Brown1985). ApoE is also synthesised by a range of other tissues, including steroidogenic organs such as the adrenal glands, testes and ovary (Blue et al. Reference Blue, Williams, Zucker, Khan and Blum1983; Polacek et al. Reference Polacek, Beckmann and Schreiber1992), lungs (Dawson et al. Reference Dawson, Lukaszewski, Ells, Malbon and Williams1989), kidney (Wallis et al. Reference Wallis, Rogne, Gill, Markham, Edge, Woods, Williamson and Humphries1983) and adipose tissue (Zechner et al. Reference Zechner, Moser, Newman, Fried and Breslow1991), and in the retinal pigment epithelial cells (Ishida et al. Reference Ishida, Bailey, Duncan, Chalkley, Burlingame, Kane and Schwartz2004).
Role of apoE in lipoprotein metabolism
ApoE is known to play a multi-functional role in lipoprotein metabolism, potentially acting as a cofactor in VLDL synthesis, the hydrolysis of VLDL remnants to produce LDL and as a high-affinity ligand for the receptor-mediated cellular removal of lipoprotein remnants. Although apoE is a constituent of Golgi VLDL, there are inconsistencies in the literature in relation to the essentiality of apoE in hepatic VLDL synthesis and secretion (Schaefer et al. Reference Schaefer, Gregg, Ghiselli, Forte, Ordovas, Zech and Brewer1986; Fazio & Yao, Reference Fazio and Yao1995; Huang et al. Reference Huang, Ji, Brecht, Rall, Taylor and Mahley1999). Undoubtedly, the most important role of apoE in lipoprotein metabolism is as a high-affinity ligand for receptors of the LDL receptor family, and the impact of genotype on lipoprotein metabolism is thought to be largely the result of an effect on the receptor binding activity of apoE. Members of this family include the LDL receptor, the LDL receptor-related protein, the VLDL receptor and the apoE receptor 2 (Strickland et al. Reference Strickland, Gonias and Argraves2002).
The apoE–receptor interactions, which mediate the cellular uptake of VLDL and chylomicron remnants, have been widely studied (Bradley & Gianturco, Reference Bradley and Gianturco1986; Mahley, Reference Mahley1988). It is thought that the basic amino acids located between residues 136 and 150, which produce a large region of positive electrostatic potential, are important for its interaction with the acidic amino acid ligand-binding region of members of the LDL receptor family (Weisgraber, Reference Weisgraber1994). Since single amino acid substitutions in this portion of the protein result in defective binding but not in complete abolition of binding activity, it is considered that the basic amino acids cooperate in the interaction with the receptor (Wilson et al. Reference Wilson, Wardell, Weisgraber, Mahley and Agard1991). Subtle changes around the LDL receptor-binding region also lead to defective receptor activity, as will be discussed.
ApoE receptor 2 (also termed LRP8) is structurally distinct from other family members in having a longer cytoplasmic domain. Furthermore, its pattern of tissue distribution is different from that of other receptors (Kim et al. Reference Kim, Iijima, Goto, Sakai, Ishii, Kim, Suzuki, Kondo, Saeki and Yamamoto1996), with apoE receptor 2 lacking in the liver but found abundantly in the brain and in several other tissues such as platelets and testes (Riddell et al. Reference Riddell, Vinogradov, Stannard, Chadwick and Owen1999). It is thought that apoE receptor 2 is involved in the role of apoE in cellular signalling pathways, which is at present poorly understood. Furthermore, the precise apoE sequence that binds to this receptor has not been established (Li et al. Reference Li, Kypreos, Zanni and Zannis2003).
ApoE also binds to scavenger receptor type BI and cell glycosaminoglycans, including heparin and heparin sulphate proteoglycans. ApoE binding to heparin sulphate proteoglycans is thought to be an initial step in the localisation of apoE-containing lipoproteins to the surface of different cell types. The best understood physiological role for this interaction is the hepatic clearance of remnant lipoproteins, contributing to the initial sequestration and subsequent uptake steps, either in association with LDL receptor-related protein or acting alone (Mahley & Ji, Reference Mahley and Ji1999; Libeu et al. Reference Libeu, Lund-Katz, Phillips, Wehrli, Hernaiz, Capila, Linhardt, Raffai, Newhouse, Zhou and Weisgraber2001).
Impact of apoE genotype on protein structure and function
In man the apoE gene is mapped to chromosome 19 in a cluster with apoC1 and apoC2. It extends for 3610 bases starting at 50 100 879 bp from pter to 50 104 489 bp from pter and consists of four exons (44, 66, 193 and 869 bp) and three introns (760, 1092 and 592 bp; Paik et al. Reference Paik, Chang, Reardon, Davies, Mahley and Taylor1985). Currently, forty-five single nucleotide polymorphisms have been identified for the apoE gene (National Center for Biotechnology Information (2006) single-nucleotide polymorphism database), twenty-seven in the intronic region and eighteen in coding regions (Table 1).
Table 1. Polymorphisms found in apoE gene exons (data from National Center for Biotechnology Information (2006) single-nucleotide polymorphism database)

SNP ID, single-nucleotide polymorphism identification; N/A, not available.
A common and widely characterised genotype is the apoE-ε missense mutations that result in three allelic isoforms ε2, ε3 and ε4 (Table 1). The protein products differ in the amino acid present at residue 112 (rs429358) and 158 (rs7412) of the protein (Tables 1 and 2). ApoE2 contains 112 Cys/158 Cys, apoE3 112 Cys/158 Arg, and apoE4 112 Arg/158 Arg (Weisgraber et al. Reference Weisgraber, Rall and Mahley1981; Rall et al. Reference Rall, Weisgraber and Mahley1982). Although the amino acids alterations do not occur within the receptor binding region (amino acids 136–150), the substitutions at positions 112 and 158 are known to impact on the salt bridge formation within the protein, which ultimately impacts on the receptor binding activity and lipoprotein ‘preference’ of the apoE protein. ApoE3 and apoE4 have comparable LDL receptors affinity, but the binding of apoE2 is 50–100 times weaker (Weisgraber et al. Reference Weisgraber, Innerarity and Mahley1982; Weisgraber, Reference Weisgraber1994). The replacement of an arginine residue with cysteine at position 158 is thought to eliminate a salt bridge between Asp154 and Arg 158 with a new bridge forming between Arg 150 and Asp 154, which dramatically alters the conformation of the receptor binding domain (Hatters et al. Reference Hatters, Peters-Libeu and Weisgraber2006; Fig. 1). The impact of genotype on the binding of apoE to other members of the LDL-receptor family is relatively unknown; although no substantial impact of isoform on LDL receptor-related protein- and VLDL receptor–apoE interactions has been observed in a series of in vitro binding studies (Ruiz et al. Reference Ruiz, Kouiavskaia, Migliorini, Robinson, Saenko, Gorlatova, Li, Lawrence, Hyman, Weisgraber and Strickland2005).
Table 2. ApoE isoform amino acid differences and physio-chemical changes
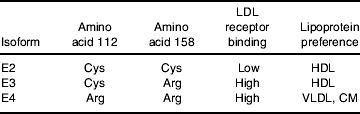
CM, chylomicrons.
The Cys112 to Arg112 substitution in apoE4, although not appearing to appreciably influence receptor binding, is thought to impact on both protein stability and carboxyl-terminal and amino-terminal domain interactions (for review, see Hatters et al. Reference Hatters, Peters-Libeu and Weisgraber2006). An arginine moiety at this position is thought to impact on the conformation of Arg61, allowing its interaction with an acidic Glu255 residue in the carboxyl-terminal (Fig. 1). This interaction affects the protein conformation, resulting in a ‘molten globule’ structure (Morrow et al. Reference Morrow, Hatters, Lu, Hochtl, Oberg, Rupp and Weisgraber2002) with a preference for larger VLDL and chylomicron remnants, in contrast to apoE2 and apoE3, which prefer smaller cholesterol-rich HDL particles. The higher lipid-binding affinity of apoE4 is not influenced by the particle size (Saito et al. Reference Saito, Dhanasekaran, Baldwin, Weisgraber, Phillips and Lund-Katz2003).
This impact on protein structure also affects molecular stability, with susceptibility of the isoforms to degradation being in the following order E4>E3>E2 (Acharya et al. Reference Acharya, Segall, Zaiou, Morrow, Weisgraber, Phillips, Lund-Katz and Snow2002).
ApoE allelic frequency and genotype distributions
Globally, the apoE allelic distribution shows substantial variation, with an allele frequency of 60–90% for the wild-type ε3 allele (Corbo & Scacchi, Reference Corbo and Scacchi1999; Singh et al. Reference Singh, Singh and Mastana2006).
The studies reviewed by Eichner et al. (Reference Eichner, Dunn, Perveen, Thompson, Stewart and Stroehla2002) demonstrate that approximately 65% of Caucasian populations are homozygous ε3/ε3, 19% are ε3/ε4, 10% are ε2/ε3, 4% are ε2/ε4, 2% are ε4/ε4 and 0·5–1% are ε2/ε2. In Europe there is a geographic cline, with 2-fold higher prevalence of the ε4 allele in northern Europe compared with southern Europe (Corbo & Scacchi, Reference Corbo and Scacchi1999; Eichner et al. Reference Eichner, Dunn, Perveen, Thompson, Stewart and Stroehla2002; Singh et al. Reference Singh, Singh and Mastana2006; Table 3), which is likely to make a contribution to the north–south differences in CVD incidence observed.
Table 3. ApoE allelic distribution in select populations worldwide (derived from Singh et al. Reference Singh, Singh and Mastana2006)

* Listed in order of ε4 allele.
† European countries.
ApoE genotype and cardiovascular risk and incidence: impact of age and gender
Over the last three decades numerous studies using a variety of CHD end points, including clinically- and angiographically-defined CHD, have investigated the impact of apoE genotype on CHD risk. The main studies have been summarised in two meta-analyses (Wilson et al. Reference Wilson, Schaefer, Larson and Ordovas1996; Song et al. Reference Song, Stampfer and Liu2004). The Wilson et al. (Reference Wilson, Schaefer, Larson and Ordovas1996) analysis summarises data from fourteen published observational studies, with carriers of the ε4 allele having an overall OR for CHD of 1·26 (95% CI 1·13, 1·41) and a non-significant OR of 0·98 (95% CI 0·85, 1·14) evident in ε2 carriers. On removing the Utermann et al. (Reference Utermann, Hardewig and Zimmer1984) study, which demonstrated a cardio-protective effect of E4 and reported results that were clearly divergent from all other studies, an OR of 1·44 (95% CI 1·27, 1·62) was observed. This finding is in agreement with the more-recent meta-analysis (Song et al. Reference Song, Stampfer and Liu2004), which includes data from 15 492 CHD cases and 32 965 controls. Overall OR of 1·42 (95% CI 1·26, 1·61) and 0·98 (95% CI 0·66, 1·46) were observed for the E4 and E2 subgroups. However, findings from the forty-eight studies included are highly heterogeneous with mean OR values derived from the individual studies ranging from 0·68 to 4·35 in ε4 carriers compared with the wild-type E3/E3 genotype. Such heterogeneity is likely to be attributable to an array of factors, including environmental factors such as smoking status and background diet, and also the age and gender of the study cohort.
Currently, a comprehensive review of the impact of age and gender on apoE genotype–CHD associations is distinctly lacking. Data from the Framingham Offspring study (Wilson et al. Reference Wilson, Myers, Larson, Ordovas, Wolf and Schaefer1994; Lahoz et al. Reference Lahoz, Schaefer, Cupples, Wilson, Levy, Osgood, Parpos, Pedro-Botet, Daly and Ordovas2001; Elosua et al. Reference Elosua, Ordovas, Cupples, Fox, Polak, Wolf, D'Agostino and O'Donnell2004) suggest a protective effect of an E2 genotype and a greater sensitivity to the deleterious effects of an E4 genotype in females compared with males. In relation to age, it appears that the impact of genotype on CVD risk is attenuated with age (Jarvik et al. Reference Jarvik, Austin, Fabsitz, Auwerx, Reed, Christian and Deeb1994; Ilveskoski et al. Reference Ilveskoski, Perola, Lehtimaki, Laippala, Savolainen and Pajarinen1999), with a lack of association of genotype with disease risk in older cohorts (Kuusisto et al. Reference Kuusisto, Mykkanen, Kervinen, Kesaniemi and Laakso1995). For example, in the Helsinki Sudden Death Study (Ilveskoski et al. Reference Ilveskoski, Perola, Lehtimaki, Laippala, Savolainen and Pajarinen1999), which conducted lesion staining of the coronary arteries of 700 individuals, age×genotype interactions were observed, with an impact of genotype only present in the group who were <53 years old.
It is speculated that the apparent age-related weakening of the association may be (a) attributable to the masking effect of an overall ‘at-risk’ phenotype, which is reflected in more extensive atherogenesis, reducing the variability and the association with any one genetic factor, or (b) because individuals who are particularly sensitive to the genotype-mediated effects may have already died and are therefore not included in the analysis of older cohorts.
ApoE genotype and physiological determinants of risk for CVD
Traditionally, an increased CVD risk in E4 carriers has been attributable to higher circulating total cholesterol and LDL-cholesterol (LDLC) in E4 carriers. As will be discussed, the sometimes moderate and often non-significantly higher circulating cholesterol levels in E4 carriers are not likely to explain the 40–50% higher CVD risk observed. Furthermore, the retention of a significant impact of genotype when correction is made for recognised lipid risk markers of disease (Terry et al. Reference Terry, Howard, Mercuri, Bond and Crouse1996; Humphries et al. Reference Humphries, Talmud, Hawe, Bolla, Day and Miller2001; Lahoz et al. Reference Lahoz, Schaefer, Cupples, Wilson, Levy, Osgood, Parpos, Pedro-Botet, Daly and Ordovas2001) suggests that the effect is partly mediated by lipid-independent mechanisms.
ApoE genotype and blood lipid levels
ApoE genotype and LDL-cholesterol levels
It has been documented that apoE genotype accounts for 7% of the variance of total cholesterol in healthy Caucasian individuals (Davignon et al. Reference Davignon, Gregg and Sing1988), and it has been suggested that an adverse cholesterol profile in E4 carriers could largely explain the increased risk of coronary events in this subgroup.
In most of the populations studied, regardless of age and health status, the ε4 allele has been associated with higher LDLC and apoB concentrations relative to E2 carriers (Table 4). However, relative to E3/E3 carriers only moderate differences in cholesterol exist, with the differences often not significant. In the studies included in Table 4 LDLC concentrations for E4 and E2 carriers are on average 8·3% higher and 14·2% lower respectively than those for E3 homozygotes, with the cholesterol-lowering effect of the ε2 allele known to be greater than the cholesterol-raising effect of ε4 allele (Davignon et al. Reference Davignon, Gregg and Sing1988; Hallman et al. Reference Hallman, Boerwinkle, Saha, Sandholzer, Menzel, Csazar and Utermann1991; Schaefer et al. Reference Schaefer, Lamon-Fava, Johnson, Ordovas, Schaefer, Castelli and Wilson1994).
Table 4. The impact of apoE genotype on LDL-cholesterol levels (E2/E4 excluded if present)

E2, E2/E2+E2/E3; E3, E3/E3; E4, E3/E4+E4/E4; MI, myocardial infarction, FHBI, familial hyperbetaproteinaemia; HRT, hormone-replacement therapy; LPG, lipoprotein glomerulopathy; N/A, not available; approx, approximately; ↑, increase; ↓, decrease.
How does apoE genotype modulate LDL-cholesterol levels?
The lower plasma LDLC in E3/E2 and E2/E2 subjects has been attributed to a number of mechanisms, including increased hepatic receptor-mediated LDL removal, lower VLDL to LDL conversion rates and decreased intestinal cholesterol absorption.
In E2 carriers defective binding of the apoE2 protein to receptors will lead to reduced hepatic VLDL and chylomicron remnant uptake, resulting in a reduced hepatic cholesterol load, which in turn will trigger up-regulation of the LDL receptor (Gregg & Brewer, Reference Gregg and Brewer1988). Increased LDL receptor expression together with reduced receptor affinity of the apoE protein would be predicted to increase apoB100-mediated LDL removal by the LDL receptor (Howard et al. Reference Howard, Gidding and Liu1998). In a number of human biokinetic studies a higher fractional catabolic rate of LDL has been observed in E2 subjects (Miettinen et al. Reference Miettinen, Gylling, Vanhanen and Ollus1992; Gylling et al. Reference Gylling, Kontula and Miettinen1995). In addition, ε2 allele carriers have been associated with lower intestinal cholesterol absorption and higher bile acid synthesis than E3 or E4 individuals (Kesaniemi et al. Reference Kesaniemi, Ehnholm and Miettinen1987; Miettinen et al. Reference Miettinen, Gylling, Vanhanen and Ollus1992; Gylling et al. Reference Gylling, Kontula and Miettinen1995). However, these results have been challenged by Von Bergman et al. (Reference Von Bergmann, Lutjohann, Lindenthal and Steinmetz2003), who have reported no differences in intestinal cholesterol absorption and synthesis in E2/E2 v. E4/E4 individuals. Also, there is currently no plausible mechanism linking apoE genotype and the efficiency of cholesterol absorption.
What about the higher LDLC levels in ε4 allele carriers? In most studies the differences relative to E3/E3 subjects are not significant, but there is a consistent trend towards higher total cholesterol and LDLC levels in E4 carriers. Although there are no differences in LDL-receptor binding between E4 and E3 individuals, as mentioned previously the amino acid change at position 112 influences the lipoprotein ‘preference’ of the protein, leading to a higher concentration associated with TAG-rich lipoproteins (chylomicrons and VLDL) as compared with E3 homozygotes (Gregg et al. Reference Gregg, Zech, Schaefer, Stark, Wilson and Brewer1986; Weisgraber, Reference Weisgraber1990). More apoE per TAG-rich lipoprotein particle would be anticipated to result in increased competition with LDL for LDL receptor-mediated clearance, which may lead to increased circulating LDLC levels (Jackson et al. Reference Jackson, Maitin, Leake, Yaqoob and Williams2006). In a number of biokinetic studies (Gregg et al. Reference Gregg, Zech, Schaefer, Stark, Wilson and Brewer1986; Demant et al. Reference Demant, Bedford, Packard and Shepherd1991; Welty et al. Reference Welty, Lichtenstein, Barrett, Jenner, Dolnikowski and Schaefer2000) a lower fractional catabolic rate of LDL-apoB100 has been reported in ε4 allele carriers. In addition, an increased conversion of VLDL to LDL-apoB100 was observed in E4 individuals. This increased synthetic rate together with the reported increased intestinal cholesterol absorption efficiency (Kesaniemi et al. Reference Kesaniemi, Ehnholm and Miettinen1987) could contribute to the trends towards higher LDLC levels in E4 carriers.
Regardless of the mechanism for the LDLC-modulating effects, it is evident that the average 8% higher LDLC levels alone cannot explain the disease differential in E4 carriers (Law et al. Reference Law, Wald and Thompson1994). Furthermore, no consistent difference in CVD risk has been observed between E2 carriers and E3/E3 individuals despite the 10–15% lower LDLC levels, which based on predictive equations would be associated with a 20–30% lower CVD risk (Law et al. Reference Law, Wald and Thompson1994). Thus, it is likely that other mechanisms in part mediate the effect of apoE genotype on CVD pathology.
ApoE genotype and other lipid risk factors for CVD
Inconsistent associations between apoE genotype and fasting TAG levels have been reported in the literature (Brown & Roberts, Reference Brown and Roberts1991; Howard et al. Reference Howard, Gidding and Liu1998; Bercedo-Sanz et al. Reference Bercedo-Sanz, Gonzalez-Lamuno, Malaga and Garcia-Fuentes1999; Inamdar et al. Reference Inamdar, Kelkar, Devasagayam and Bapat2000; Szalai et al. Reference Szalai, Czinner and Csaszar2000; Tan et al. Reference Tan, Tai, Tan, Chia, Lee, Chew and Ordovas2003), and a meta-analysis (Dallongeville et al. Reference Dallongeville, Lussier-Cacan and Davignon1992) has concluded that E2/E2, E2/E4 E2/E3 and E3/E4 subgroups have higher fasting TAG levels than E3/E3 individuals. Higher fasting TAG levels are thought to be attributable to the limited receptor affinity of the apoE2 protein present on VLDL remnants resulting in impaired hepatic clearance of TAG-rich lipoproteins. The mechanisms that could potentially contribute to the moderate hypertriacylglycerolaemia evident in E4 carriers are currently unclear.
Plasma TAG levels in the postprandial state (postprandial lipaemia) are recognised to be a stronger determinant of CVD risk relative to fasting TAG levels (Zilversmit, Reference Zilversmit1979; Patsch et al. Reference Patsch, Esterbauer, Foger and Patsch2000). It has been shown (Weintraub et al. Reference Weintraub, Eisenberg and Breslow1987; Dallongeville et al. Reference Dallongeville, Tiret, Visvikis, O'Reilly, Saava, Tsitouris, Rosseneu, DeBacker, Humphries and Beisiegel1999) that E2 carriers have a relatively delayed exaggerated chylomicron remnant clearance and exaggerated lipaemia and a homozygous E2/E2 genotype is one of the recognised causes of a type III hyperbetalipoproteinaemia phenotype. However, the majority of studies (Brenninkmeijer et al. Reference Brenninkmeijer, Stuyt, Demacker, Stalenhoef and van't Laar1987; Weintraub et al. Reference Weintraub, Eisenberg and Breslow1987; Brown & Roberts, Reference Brown and Roberts1991; Boerwinkle et al. Reference Boerwinkle, Brown, Sharrett, Heiss and Patsch1994; Orth et al. Reference Orth, Wahl, Hanisch, Friedrich, Wieland and Luley1996) show that only E2 homozygotes have impaired chylomicron remnant clearance, with one ε3 allele largely compensating for the impaired receptor binding. As for the implication of the ε4 allele in postprandial metabolism, the data published are inconsistent, with only moderate trends towards impaired metabolism observed.
Although apoE is a constituent of HDL, there are much less data available on the effect of apoE genotype on HDL-cholesterol (HDLC) than there are on LDLC levels. In general, there is a trend towards a reduction in circulating HDLC levels from an E2 genotype to an E4 genotype; some studies (Dallongeville et al. Reference Dallongeville, Lussier-Cacan and Davignon1992; Howard et al. Reference Howard, Gidding and Liu1998; Dallongeville et al. Reference Dallongeville, Tiret, Visvikis, O'Reilly, Saava, Tsitouris, Rosseneu, DeBacker, Humphries and Beisiegel1999; Minihane et al. Reference Minihane, Khan, Leigh-Firbank, Talmud, Wright, Murphy, Griffin and Williams2000; Tan et al. Reference Tan, Tai, Tan, Chia, Lee, Chew and Ordovas2003) have reported effects of genotype on HDLC and apoA1 levels, while other studies (Bercedo-Sanz et al. Reference Bercedo-Sanz, Gonzalez-Lamuno, Malaga and Garcia-Fuentes1999; Szalai et al. Reference Szalai, Csaszar, Czinner, Palicz, Halmos and Romics1999; Inamdar et al. Reference Inamdar, Kelkar, Devasagayam and Bapat2000; Sheehan et al. Reference Sheehan, Bennett and Cashman2000) have not demonstrated an association. As fasting TAG levels are known to be an important determinant of HDL metabolism and HDLC levels, it may be predicted that lower HDLC levels may be evident in E2 and E4 carriers. This lack of TAG–HDLC response to genotype suggests that a TAG-independent mechanism may also play a role in modulating the effect of genotype on HDLC.
ApoE genotype and responsiveness to dietary fat manipulation
The influence of environmental factors on genotype–disease associations is being increasingly recognised. A limited number of studies have indicated that alcohol intake influences apoE–CVD associations (Corella et al. Reference Corella, Guillen, Saiz, Portoles, Sabater, Cortina, Folch, Gonzalez and Ordovas2001a,Reference Corella, Tucker, Lahoz, Coltell, Cupples, Wilson, Schaefer and Ordovasb), but the greatest evidence exists for an impact of smoking status and dietary total fat content and fatty acid composition on the LDLC modulatory effects of the apoE genotype. Responsiveness to dietary fat manipulation is recognised to be highly variable, with genetic variability known to be partly responsible. The systematic review by Masson et al. (Reference Masson, McNeill and Avenell2003) includes studies that have examined the impact of genotype on the responsiveness of fasting lipids to dietary cholesterol (fifteen individual studies) and total fat or fatty acid composition (thirty-six individual studies; mainly manipulation of SFA, MUFA and PUFA ratios). Three of the cholesterol-manipulation studies have reported a greater circulating cholesterol response in E4 carriers. Eleven of the studies that manipulated dietary fat have demonstrated a genotype×treatment interaction, with the E4 subgroup being generally the most responsive (Masson et al. Reference Masson, McNeill and Avenell2003). For example, in the Schaefer et al. (Reference Schaefer, Lamon-Fava, Ausman, Ordovas, Clevidence, Judd, Goldin, Woods, Gorbach and Lichtenstein1997) study (n 148) a National Cholesterol Education Program Step 2 diet was found to result in an overall mean reduction in LDLC levels of 19% and 16% in men and women respectively, with corresponding response ranges of +3% to –55% and +13% to –39%. In the male participants, but not in the female participants, an E4 genotype was shown to be associated with greater LDLC reductions. In the systematic review (Masson et al. Reference Masson, McNeill and Avenell2003) the lack of significance reported in many of the studies is likely to be attributable to a lack of power to detect an inter-genotype difference in response, rather than a lack of a ‘real’ biological effect of apoE genotype. Many of the studies included cohorts of less than fifty participants and retrospective apoE genotype profiling, which often resulted in small group sizes in the rare allele groups. Of the eleven studies that reported significant impacts of apoE genotype, six included more than fifty participants, with an additional study that included forty-five participants (n 15 for E3/E3, E3/E4 and E4/E4 groups) prospectively recruiting on the basis of apoE genotype (Sarkkinen et al. Reference Sarkkinen, Korhonen, Erkkila, Ebeling and Uusitupa1998).
It is likely that background dietary fat composition is partly responsible for the variation in associations between apoE genotype and CVD risk and blood lipid profile reported in the literature. Furthermore, in E4 individuals with a high-fat high-cholesterol high-SFA diet dietary fat manipulation may offer a viable means of counteracting the increased CVD risk. However, before this approach can be advocated with any certainty additional adequately-powered studies are needed in order to fully elucidate the impact of apoE genotype on the heterogeneity in response to dietary total fat and SFA, MUFA and PUFA content.
Recent evidence (Minihane et al. Reference Minihane, Khan, Leigh-Firbank, Talmud, Wright, Murphy, Griffin and Williams2000) also suggests that apoE genotype may in part predict the LDLC response to fish oil fatty acid intervention. The variability of LDLC-raising effect of EPA and DHA has been frequently reported (Harris, Reference Harris1997). In a study of individuals with an atherogenic lipoprotein phenotype (Minihane et al. Reference Minihane, Khan, Leigh-Firbank, Talmud, Wright, Murphy, Griffin and Williams2000) retrospective genotyping suggests that the LDLC-raising effects observed following supplementation with 3 g EPA+DHA/d are associated with an apoE4 genotype. Additional studies are currently underway to investigate EPA/DHA–LDLC associations.
Lipoprotein-independent effects of the apoE protein and apoE genotype: impact on macrophage, endothelial cell, smooth muscle cell and platelet function
As mentioned earlier, although an E4 genotype is associated with moderately-higher LDLC and TAG levels and a trend towards lower HDLC levels, these effects alone are unlikely to be responsible for the higher CVD burden, even in individuals with a high total fat and saturated fat intake. It is therefore speculated that lipid-independent mechanisms may contribute substantially to disease risk.
Monocyte-derived macrophages can produce up to 20% of the total apoE (Basu et al. Reference Basu, Brown, Ho, Havel and Goldstein1981, Reference Basu, Ho, Brown, Bilheimer, Anderson and Goldstein1982; Newman et al. Reference Newman, Dawson, Rudel and Williams1985; Wang-Iverson et al. Reference Wang-Iverson, Gibson and Brown1985). The anti-atherogenic roles of macrophage apoE have been demonstrated in apoE-null rodents (Bellosta et al. Reference Bellosta, Mahley, Sanan, Murata, Newland, Taylor and Pitas1995; Thorngate et al. Reference Thorngate, Rudel, Walzem and Williams2000). In these animals low-level tissue-specific expression of human apoE in macrophages inhibits atherogenesis without substantially influencing the plasma lipid profile.
The role of locally-secreted apoE in the artery wall is currently only partly understood, but it has been proposed to exert several biological functions (Fig. 2). Acting as a paracrine agent, macrophage-derived apoE is known to influence smooth muscle cell (Swertfeger & Hui, Reference Swertfeger and Hui2001), endothelial cell (Stannard et al. Reference Stannard, Riddell, Sacre, Tagalakis, Langer, von Eckardstein, Cullen, Athanasopoulos, Dickson and Owen2001), lymphocyte (Mistry et al. Reference Mistry, Clay, Kelly, Steiner and Harmony1995) and platelet (Riddell et al. Reference Riddell, Graham and Owen1997) function. Within the macrophage itself apoE is involved in reverse cholesterol efflux from macrophages (Shimano et al. Reference Shimano, Ohsuga, Shimada, Namba, Gotoda, Harada, Katsuki, Yazaki and Yamada1995) and is known to modulate the cell inflammatory response through an impact on NO and proinflammatory cytokine production (Colton et al. Reference Colton, Czapiga, Snell-Callanan, Chernyshev and Vitek2001, Reference Colton, Brown, Cook, Needham, Xu, Czapiga, Saunders, Schmechel, Rasheed and Vitek2002). Although data is currently lacking, accumulating evidence suggests an impact of apoE genotype on these metabolic processes, which may be attributable partly to differences in the antioxidant capacity of the apoE isoforms.

Fig. 2. Local effects of apoE on the artery wall. M, monocyte; MΦ, macrophage; EC, endothelial cell; P, platelet; T, T lymphocyte; SMC, smooth muscle cells; VCAM-1, vascular cell adhesion molecule-1.
ApoE and platelet aggregation
Desai et al. (Reference Desai, Bruckdorfer, Hutton and Owen1989) have observed that the binding of apoE as a component of large HDL2 particles to saturable sites on platelets is associated with an inhibition of platelet aggregation. More recent studies (Riddell et al. Reference Riddell, Graham and Owen1997, Reference Riddell, Vinogradov, Stannard, Chadwick and Owen1999, Reference Riddell, Sun, Stannard, Soutar and Owen2001) have suggested that apoE may inhibit platelet reactivity by interacting with apoE receptor 2, which would result in an increase in cellular NO levels as a result of simulation of the NO synthase signalling cascade. The impact of apoE genotype on the anti-aggregatory effect of apoE has not been investigated.
ApoE and adhesion molecule expression
In endothelial cells the interaction of apoE with apoE receptor 2 has been proposed to activate NO synthase through an effect on 1-phosphatidylinositol 3-kinase signalling, with a resultant NO-induced inhibition of vascular cell adhesion molecule-1 induction (Stannard et al. Reference Stannard, Riddell, Sacre, Tagalakis, Langer, von Eckardstein, Cullen, Athanasopoulos, Dickson and Owen2001). In a cell-culture model (EAhy926) Sacre et al. (Reference Sacre, Stannard and Owen2003) have observed an isoform-specific induction of endothelial NO in the order E3>E2>E4. The impact of apoE genotype on adhesion molecule expression in vivo is unknown, although a recently-completed study (AM Minihane et al. unpublished results) indicates an effect of apoE genotype on circulating vascular cell adhesion molecule levels in human volunteers, with the relative levels (E4>E3>E2) consistent with the NO induction observed by Sacre et al. (Reference Sacre, Stannard and Owen2003).
ApoE and smooth muscle cell migration and proliferation
Smooth muscle cell migration into the intima and subsequent proliferation are considered to play an important role in atherosclerosis. ApoE has been shown to inhibit platelet-derived growth factor-directed smooth muscle cell migration by binding to LDL receptor-related protein, which activates the cAMP, protein kinase cascade (Hui & Basford, Reference Hui and Basford2005). In addition, apoE inhibits cell proliferation through binding to cell surface proteoglycans, by a mechanism in which inducible NO synthase is increased (Hui & Basford, Reference Hui and Basford2005). It has been demonstrated that the isoforms do not differ in terms of cell migration inhibition, since binding of lipid-free apoE to LDL receptor-related protein does not show isoform preferences (Zeleny et al. Reference Zeleny, Swertfeger, Weisgraber and Hui2002). On the contrary, apoE2 and apoE3 are more efficient in inhibiting smooth muscle cell proliferation than apoE4 (Zeleny et al. Reference Zeleny, Swertfeger, Weisgraber and Hui2002), which is consistent with the different binding capacity of apoE to heparin sulphate proteoglycans (Cullen et al. Reference Cullen, Cignarella, Brennhausen, Mohr, Assmann and von Eckardstein1998; Hara et al. Reference Hara, Matsushima, Satoh, Iso-o, Noto, Togo, Kimura, Hashimoto and Tsukamoto2003).
ApoE and cellular cholesterol efflux and reverse cholesterol transport
The involvement of apoE in mediating cholesterol efflux from macrophages was first identified by Basu et al. (Reference Basu, Ho, Brown, Bilheimer, Anderson and Goldstein1982) and is now supported by several lines of evidence (Shimano et al. Reference Shimano, Ohsuga, Shimada, Namba, Gotoda, Harada, Katsuki, Yazaki and Yamada1995).
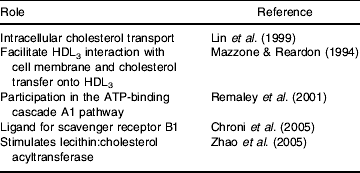
ApoE seems to promote cholesterol efflux when endogenously expressed and to a lesser extent when exogenously added (Lin et al. Reference Lin, Duan and Mazzone1999), and it has been hypothesised that the macrophage and non-macrophage apoE act via divergent mechanisms (Lin et al. Reference Lin, Duan and Mazzone1999) that work in parallel (Dove et al. Reference Dove, Linton and Fazio2005). The enhancing effect can be observed in the absence of acceptors (Zhang et al. Reference Zhang, Gaynor and Kruth1996) and in the presence of cholesterol acceptors such as HDL or phospholipid vesicles (Mazzone & Reardon, Reference Mazzone and Reardon1994). There is a very complex literature relating to the mechanisms by which apoE influences cholesterol efflux in macrophages, and several mechanisms have been proposed (Table 5).
Table 5. Proposed roles for apoE in reverse cholesterol transport
The metabolism of cholesterol in macrophages has been found to differ among the three isoforms. In the absence of extracellular acceptors cholesterol-loaded monocyte-derived macrophages isolated from E4/E4 carriers are less effective in cholesterol efflux than E3/E3 cells, which are less effective than E2/E2 cells (Cullen et al. Reference Cullen, Cignarella, Brennhausen, Mohr, Assmann and von Eckardstein1998). In mouse macrophages (RAW 264·7) the efficiency of cholesterol efflux is in the order E2>E3>E4, which is attributed to isoform variations in binding capacities to heparin sulphate proteoglycans. A higher binding activity of apoE4 is considered to result in higher uptake or degradation of apoE, which results in lower cholesterol efflux activity (Hara et al. Reference Hara, Matsushima, Satoh, Iso-o, Noto, Togo, Kimura, Hashimoto and Tsukamoto2003). This lower efficiency of cholesterol efflux in E4 individuals could make an important contribution to the higher CVD burden observed.
ApoE, NO production and inflammatory status
NO is regarded as a potent macrophage pro-inflammatory mediator. The addition of apoE has been shown to increase monocyte-derived macrophage NO production (Colton et al. Reference Colton, Czapiga, Snell-Callanan, Chernyshev and Vitek2001) by increasing the uptake of arginine (the substrate for NO production) as a result of the up-regulation of the cationic acid transporter family (Colton et al. Reference Colton, Czapiga, Snell-Callanan, Chernyshev and Vitek2001). ApoE isoform-mediated differences in monocyte-derived macrophage NO production have been observed in several models, with higher levels of NO produced by apoE4 macrophages compared with apoE3 macrophages (Colton et al. Reference Colton, Needham, Brown, Cook, Rasheed, Burke, Strittmatter, Schmechel and Vitek2004).
In addition to NO, macrophages produce and secrete an array of pro-inflammatory cytokines, including a number of chemokines, which impact on atherogenesis in both an autocrine and paracrine manner. Data on the impact of apoE genotype on the macrophage inflammatory response are very limited. In a recent studies by Ophir et al. (Reference Ophir, Meilin, Efrati, Chapman, Karussis, Roses and Michaelson2003, Reference Ophir, Amariglio, Jacob-Hirsch, Elkon, Rechavi and Michaelson2005) and Lynch et al. (Reference Lynch, Tang, Wang, Vitek, Bennett, Sullivan, Warner and Laskowitz2003) higher production of pro-inflammatory cytokines in the brain and serum was observed in E4 v. E3 transgenic mice following injection with lipopolysaccharide (inflammatory stimulus). The study of Lynch et al. (Reference Lynch, Tang, Wang, Vitek, Bennett, Sullivan, Warner and Laskowitz2003) highlights that the impact of genotype is largely attributable to a differential impact of E3 v. E4 on NF-κB signalling, which may be attributable to apoE genotype-mediated differences in oxidative status.
ApoE genotype and oxidative status
There are several lines of evidence demonstrating that apoE has antioxidant capacity (Hayek et al. Reference Hayek, Oiknine, Brook and Aviram1994; Pratico et al. Reference Pratico, Lee, Trojanowski, Rokach and Fitzgerald1998; Aviram et al. Reference Aviram, Dornfeld, Rosenblat, Volkova, Kaplan, Coleman, Hayek, Presser and Fuhrman2000; Kitagawa et al. Reference Kitagawa, Matsumoto, Kuwabara, Takasawa, Tanaka, Sasaki, Matsushita, Ohtsuki, Yanagihara and Hori2002). Miyata & Smith (Reference Miyata and Smith1996), whilst investigating the impact of apoE genotype on Alzheimer's disease pathology, were the first to propose allele-specific differences in the antioxidant capacities of apoE isoforms in the order E2>E3>E4, with E2 emerging in in vitro systems as having a 2-fold higher antioxidant capacity relative to E4. Subsequent in vitro studies and brain autopsy investigations of patients with Alzheimer's disease (Jolivalt et al. Reference Jolivalt, Leininger-Muller, Bertrand, Herber, Christen and Siest2000; Tamaoka et al. Reference Tamaoka, Miyatake, Matsuno, Ishii, Nagase and Sahara2000) have confirmed these earlier findings.
Indirect but strong evidence for a role of apoE-mediated differences in oxidative stress being important in CVD pathology is provided by two recent prospective cardiovascular surveillance studies, i.e. the Northwick Park Heart Study (Humphries et al. Reference Humphries, Talmud, Hawe, Bolla, Day and Miller2001) and the Framingham Offspring Study (Talmud et al. Reference Talmud, Stephens, Hawe, Demissie, Cupples, Hurel, Humphries and Ordovas2005). Both studies conclude that after correction for classical risk factors (including lipids) an increased risk of CVD in E4 carriers is only evident in those who smoke, which strongly indicates that an impact of genotype on oxidative status is important. The results of the Northwick Park Heart Study are presented in Table 6, with an adjusted (including for blood lipids) hazard ratio of 2·79 in E4 carriers who were smokers compared with a combined genotype non-smoking group. Although no data is currently available, based on the smoking–genotype interaction observed it may also be speculated that the impact of an E4 genotype may be more evident in individuals with a low dietary antioxidant intake.
Table 6. CHD adjusted hazard ratios (HR) according to apoE genotype for men participating in the Northwick Park Heart StudyFootnote * (adapted from Humphries et al. Reference Humphries, Talmud, Hawe, Bolla, Day and Miller2001)

E2 carriers, E2/E2, E2/E3; E4 carriers, E3/E4, E4/E2.
* Results are compared with the never-smokers, all genotypes combined.
† Results adjusted for clinic, age, BMI, systolic blood pressure, plasma lipids (cholesterol and TAG) and fibrinogen.
Recent evidence (Dietrich et al. Reference Dietrich, Hua, Block, Olano, Packer, Morrow, Hudes, Abdukeyum, Rimbach and Minihane2005; Jofre-Monseny et al. Reference Jofre-Monseny, de Pascual-Teresa, Plonka, Huebbe, Boesch-Saasatmandi, Minihane and Rimbach2007) supports a role of apoE genotype in mediating oxidative status. In a mixed smoking and non-smoking group 29% higher levels of lipid peroxidation (as measured by circulating F2-isoprostane levels) were observed in individuals with a total plasma cholesterol >5·6 mmol/l (Dietrich et al. Reference Dietrich, Hua, Block, Olano, Packer, Morrow, Hudes, Abdukeyum, Rimbach and Minihane2005). Furthermore, in a murine macrophage (RAW 264.7) cell line stably transfected with the human apoE3 and apoE4 gene it was observed that an apoE4 genotype is associated with increased membrane oxidation and NO and superoxide anion radical production (Jofre-Monseny et al. Reference Jofre-Monseny, de Pascual-Teresa, Plonka, Huebbe, Boesch-Saasatmandi, Minihane and Rimbach2007).
The exact molecular mechanism by which apoE could exert its antioxidant effects and why it is isoform-dependent is not well understood. A number of possible mechanisms have been suggested, including an effect of genotype on protein folding impacting on the metal-binding domain of the protein located in the amino terminal (Miyata & Smith, Reference Miyata and Smith1996; Pham et al. Reference Pham, Kodvawala and Hui2005). Whatever the mechanism, it seems likely that genotype differences in oxidative status, in particular within the microenvironment of the arterial intima, are partly responsible for the higher CVD risk in E4 carriers, and that therapies targeted at reducing oxidative status and its metabolic consequences could help negate the deleterious effects of an apoE genotype.
Conclusion
Although extensively investigated, the role of the apoE protein and the impact of apoE genotype on cardiovascular health and pathology are only partly understood. It is now evident that part of the CVD burden associated with an E4 genotype is independent of an effect on lipoprotein metabolism, with an impact of genotype on oxidative status and macrophage function being increasingly recognised. Furthermore, observational and intervention trials based on retrospective genotyping of the study participants have highlighted the impact of environmental factors such as smoking status and dietary fat composition on genotype–phenotype associations. Further studies using a large-scale retrospective-genotyping approach or a smaller more-focused approach with individuals prospectively recruited on the basis of genotype are needed to establish the potential of different dietary manipulations to counteract the increased CVD risk in E4 carriers (25% of the UK population). However, it is recognised that because of the complexity and cost such an approach cannot be used to investigate all potential genotype–environment–phenotype associations. Human transgenic cells and animal models can provide a useful tool to initially screen potential dietary components of interest.








