


INTRODUCTION
Information on the rate of new infections is critical to monitoring the HIV pandemic and to measuring the impact of prevention interventions [Reference Brookmeyer and Quinn1, Reference Busch2]. Over 15 years ago, Brookmeyer & Quinn [Reference Brookmeyer and Quinn1] and Janssen et al. [Reference Janssen3] introduced the idea that HIV incidence could be reliably measured by conducting cross-sectional surveys and counting the number of individuals with ‘incident’ infections detected by laboratory assays. This method was proposed to overcome the expense and bias associated with longitudinal cohort studies and the complexities of mathematical modelling. However, incidence estimation in cross-sectional surveys depends on being able to distinguish individuals who have recent infection (i.e. ideally those acquiring infection in the last 12 months) from those with longstanding infection (>12 months).
However, a host of factors have adversely affected the performance of HIV incidence assays, resulting in a substantial number of ‘false recent’ infection results among individuals who actually have longstanding HIV infections; the rate of such results can vary markedly between populations and over time. These factors include variability in immune responses at both an individual and population-level, variability by HIV-1 subtype, access to antiretroviral therapy (ART), advanced HIV disease, and other factors that are not well understood. Notably, the inability to account for these factors when estimating incidence, especially from early generation assays, has led to unreliable estimates of the number and pattern of new infections in some countries. These limitations, as well as the limited period for which some assays can identify recent infection, present significant barriers to the development and application of HIV incidence assays.
In 2011, stakeholders convened as an Incidence Assay Critical Path Working Group, to review challenges and propose ways to improve assay development especially for use in a surveillance and research context [4]. To help overcome these challenges, the Bill and Melinda Gates Foundation (BMGF) awarded a grant to form the Consortium for the Evaluation and Performance of HIV Incidence Assays (CEPHIA). CEPHIA was tasked with: providing clear guidelines for cross-sectional incidence estimation; fostering scientific consensus through defining a target product profile for an assay designed to assess incidence at a population level; identifying development priorities and performance metrics for incidence assays; and establishing a repository of specimens to assess the most promising available incidence assays and enable development and rigorous evaluations of multi-test algorithms for incidence measurement. Creating this framework has significantly reshaped the landscape for developing and evaluating HIV incidence assays.
In this paper, we review these developments and describe the remaining challenges to establishing reliable tools, based on HIV recency assays, for cross-sectional HIV incidence estimation at the population level. To date, about 20 assays have been developed or adapted to estimate HIV incidence. These assays work by using markers of the maturity of immune response to classify HIV-seropositive specimens as belonging to either a recently or non-recently infected person. Until 2012, only one dedicated incidence assay was commercially available (BED) [Reference Parekh5]; at that time a second dedicated assay, the Sedia™ HIV-1 LAg-Avidity assay, was commercially released [Reference Duong6]. Most of the remaining assays now available are modified commercial HIV diagnostic assays. Table 1 identifies the major assays currently available, as well as historically important assays.
Table 1. Historically important and currently available HIV incidence assays
Multiple strategies are now used to improve incidence estimation efforts. For example, a combination of antibody-based assays and viral load has led to improvements in the accuracy of HIV incidence estimation, reducing the false recency ratio (FRR) of these assays by mitigating the measurement error resulting from patients on ART. Building on this idea is the concept of a recent infection testing algorithm (RITA), which uses a series of assays in combination – often an HIV screening test, and antibody-based recency assay, and a viral load assay. RITAs were recently used in household surveys in Kenya [Reference Kimanga7], Botswana and South Africa [Reference Rehle8]. Starting in 2015, the United States Government has been investing in population-based HIV impact assessment surveys (PHIAs) that use RITAs to measure the impact of HIV prevention programmes in about 20 sub-Saharan African countries. It is important to have consensus on approaches to estimating incidence using RITAs, and to understand the implications of RITA performance on study design and analysis, to ensure that accurate and informative incidence estimates are generated.
To promote consistency in estimating HIV incidence using RITAs at the population level, the WHO Working Group on HIV Incidence Assays published guidelines in 2010 and have provided technical updates in 2013 and 2015 [9, 10], with a further technical update planned for 2016. These guidelines reflect the most up to date recommendations for using HIV incidence assays for surveillance and epidemic monitoring and have also been used to inform a revision of the 2005 Guidelines for Estimating National HIV Prevalence published in 2015 by the UNAIDS/WHO HIV Surveillance Global Working Group to address the use of HIV incidence assays in population surveys.
NEW RESOURCES FOR DEVELOPERS AND USERS OF INCIDENCE ASSAYS
Defining performance metrics, refining statistical tools.
The methods for calculating incidence estimates have been refined using laboratory assessment of recent infection biomarkers [Reference Hargrove11–Reference Kassanjee19]. The critical constructs, on which this characterization of recency tests rest, are:
-
• The protocol dependent, estimated date of detectable infection. The specific assays chosen for use in any particular HIV infection screening protocol will have an impact on when infection becomes detectable, and, consequently, the calculation of the estimation of this date of detectable infection. All assays have their own median delay of days, from initial HIV exposure/acquisition until infection becomes detectable, which affects this calculation. It is important to be explicit about this largely neglected ‘front end’ of the case definition of ‘recent infection’. It is further necessary to account for ‘NAAT yield’ (i.e. specimens that test positive for HIV nucleic acid markers, but prior to HIV antibody seroconversion thus making the antibody-based recency assays unusable) in the case definition of ‘recent infections’. These NAAT yield positive specimens offer an increased mean duration of recent infection with no significant increase in false recency, as long as the diagnostic algorithm is highly specific (false positives are likely to be classified as recent on most existing incidence assays).
-
• Recency cut-off period (T). To determine whether an assay is producing false recent results, a time boundary for recency must be defined. Using this pre-defined T, a test result of ‘recent’ obtained from a specimen whose subject was known to have had detectable infection for a time longer than T can be properly defined as a false recent result. Current WHO recommendations are for T to be set at 2 years (http://www.unaids.org/sites/default/files/media_asset/HIVincidenceassayssurveillancemonitoring_en.pdf). Given the lack of a gold standard for ‘recent’ infection and the approximate nature of most estimated dates of infection, it is challenging to decide on an appropriate recency ‘cut-off’ time (T). Ultimately, the period of recency need not be defined such that assays can be calibrated to shift from ‘recent’ to ‘longstanding’ results at precisely T; rather, the decision regarding the value of T simply requires that the period of recency be sufficiently long lasting so that surveys of feasible size can expect to detect a statistically stable number of recent infections, yet not so long that surveys are reduced to essentially estimating prevalence only. Kassanjee et al. [Reference Kassanjee13] highlight a consistent method to estimate incidence from cross-sectional surveys using a recency cut-off beyond which a known incorrect ‘recent’ test result can be reclassified – using this method, cut-off T is variable based on particular situations, and is sensibly chosen so that the FRR (defined below) for the test is very small.
-
• Mean duration of recent infection (MDRI). This is the mean time that a group of subjects fit the ‘recent’ case definition, after initial detectable infection and within the period T. Note that this does not require that progression from recent to non-recent is a once-off transition. Therefore, MDRI calculations consistently account for both inter-subject variability in biomarker development and intra-subject fluctuations, both of which are likely.
-
• False recency ratio (FRR). This is simply the proportion of those who are classified as recent by the test, despite being known to be infected for more than time T. The FRR is context-dependent, because populations of interest have differing proportions of elite controllers, antiretroviral-treated virally suppressed persons, and other characteristics less well understood that are likely to produce a recent false test, such as regional variation in prevalent HIV subtype and host genetic factors. Naturally, an ideal test would have an FRR of 0% in all contexts, but in practice there will always be residual uncertainty about the FRR. When the FRR is shown to be sufficiently close to zero, biomarker-based incidence estimates will have a greater level of precision.
Both in assay development and field application, the concept of estimated date of detectable infection must be carefully considered:
-
• Estimated date of detectable infection (EDDI) of a particular subject in a study. In practice, an EDDI is a summary measure of uncertain infection time (essentially a ‘plausibility window’) derived from earliest plausible and last plausible dates of detectable infection, obtained by interpreting diagnostic testing histories (further refinement is possible by incorporating uncertainty in the ‘diagnostic delays’ of the relevant tests). A systematic measure of EDDIs for individual subjects is necessary, as it is almost inevitable that in a large set of specimens analysed together, specimens would be obtained from a range of different testing protocols; all then need to be consistently processed when calculating a single estimate such as MDRI. We propose that it would be useful to standardize the definition of ‘detectable infection’ as the date at which a subject would first yield a detectable viral load on a highly sensitive viral load assay, i.e. with a detection threshold of 1 copy per ml.
-
• Use of an MDRI offset related to the survey test protocol used. In real-world testing protocols, infections become detectable at different times post exposure depending on the assays used in the particular protocol. The actual screening procedure determines the selection of specimens reflexed to the recency-testing protocol, or meeting the case definition of ‘recent’ without further testing. For this reason, the MDRI must always be adapted to the sensitivity of the actual screening procedure. It will seldom, if ever, be possible to find a study-specific MDRI in a table of values calculated by a test development or test benchmarking study.
The challenge for a developer of an incidence assay is to maximise the MDRI (hence having more recent cases to count in a survey, thus improving statistical precision and power at a given sample size) while keeping the FRR low (reducing the measurement error within recency data resulting from misclassified longstanding infections). By specifying an intended-use context and interpreting the impact of test properties on the precision of the incidence estimate in that context, this trade-off between MDRI and FRR can be made precise [Reference Kassanjee13] . While there are already recent infection case definitions with a FRR around 1% in likely real-world contexts, existing tests currently obtain an MDRI of only a few months (Table 4). MDRIs of close to 12 months are needed to have usefully precise incidence estimates in the order of 1% or 2% per annum, using samples of a few thousand individuals.
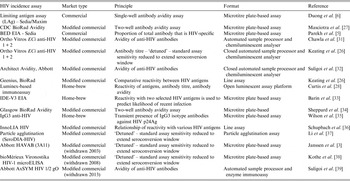
To demonstrate this, Figure 1 displays the impact that MDRI and FRR have on the real-world application of these assays, using prevalence and expected incidence for Botswana as an example. The figure shows the sample sizes required to support a statistically significant comparison between two incidence measurements where the second measurement is expected to be at least 40% lower than the first, plotted as a function of MDRI for several different values of FRR. In addition to the MDRI and FRR (which are in turn impacted by variables such as HIV-1 subtype, extent of ART use and viral load suppression in the population being surveyed), the sample size is also dependent on factors related to the nature of the epidemic (i.e. prevalence and expected incidence), the survey methods (and consequent requirement to include design effects), and the desired level of precision associated with the incidence estimate. For example, in countries with higher incidence than Botswana, the required sample sizes would be lower since the expected number of events detected will be higher. Conversely, if the background prevalence is higher, the required sample size will also be higher, since the proportion of HIV-positive individuals identified in the survey who are recently infected is lower. Given the performance characteristics of currently available incidence assays, detection of a decrease in incidence of ⩾40% at the national level can be performed using a sample size of ⩽20 000 for only two countries (Lesotho and Swaziland), while assay-based estimation of incidence at a single point in time with a relative standard error of 30% can be achieved in nearly all of the 14 countries with prevalence of at least 5% or incidence of at least 0·3% per annum, as recommended by WHO and UNAIDS [10]. However there is significant uncertainty regarding the MDRI and FRR assumptions in countries where multiple subtypes are known to be circulating, such as Kenya, Tanzania and Uganda (subtypes A and D) and Cameroon (circulating recombinant form CRF02_AG).
Fig. 1. Impact of mean duration of recent infection and false recency ratio on sample size requirements to detect reductions in incidence. This figure demonstrates the sample size required to detect a 40% reduction in incidence for the country of Botswana (power 0·8, alpha 0·05, design effect 1·3). Calculations come from https://finddx.shinyapps.io/Sample_size_calculator/.
In general, to define and estimate the performance of a recent infection test, one must first specify contextual factors (such as incidence, prevalence, treatment coverage) and details of intended use. Then, various conventional metrics such as power and precision can be calculated from these inputs, and potentially be optimised as a function of controllable inputs such as choice of test, or the threshold applied to the test to define as ‘recent’.
Target product profile (TPP)
Determination of whether or not a particular diagnostic test is suitable for a specific application is typically achieved by comparing its performance characteristics to a TPP. In 2011, a TPP for incidence assays was described to be applied to the estimation of incidence in a population, based on the experience of users of early HIV incidence assays, laboratorians, epidemiologists and regulatory agencies [4]. The TPP includes minimal acceptable recommendations for MDRI (at least 120 days) and FRR (<2%). The TPP also requires that assay results be reproducible, and the training, equipment and sample type requirements be practical for the populations to be studied and location where testing will be performed. Additionally, the TPP addresses practical requirements of a test such as reagent storage conditions, sample testing volume and conditions, training, and infrastructure needs. TPP criteria related to practicality of use were included to ensure that evaluation of an assay takes into account the needs of resource-limited settings where complex or costly automated analysers may not be available.
In addition to incidence estimation in a population, recent infection tests could also be used for other purposes. Alternative use cases include assessment of the impact of large-scale HIV prevention interventions, infection staging to guide individual treatment or public health interventions (e.g. contact tracing), or case-based surveillance by central laboratories where additional types of data (e.g. CD4+ T-cell counts and viral loads) may be available. Thus, additional TPPs may be required that correspond to other use cases (see Table 2 for a summary of potential use cases for HIV incidence assays).
Table 2. Potential uses for HIV incidence assays

The expansion of use cases and TPPs for HIV incidence assays may also impact the market size. In 2010, a market landscape assessment was performed to describe the projected demand for incidence assays under several different scenarios (http://www.who.int/diagnostics_laboratory/links/assays_to_estimate_hiv_incidence_jun_2010.pdf). An updated market landscape assessment has been commissioned to provide up-to-date information on the potential market for these assays, taking into account the potential for wider use, including on an individual patient basis.
Defining an HIV incidence assay development critical path
The critical path for an incidence assay depends on its position on the spectrum ranging from early biomarker discovery to post-marketing performance evaluation. Figure 2 illustrates some of the distinct challenges facing developers at each stage of assay discovery and development; each of these challenges requires specific types of resources. Historically, the focus of HIV incidence assays was on the maturation of the humoral immune response. However, it is increasingly clear that the assays that are likely to be most useful in the next decade are only now being developed. New approaches include rapid tests for incidence and numerous non-traditional approaches, which are being developed with the hope they may not be susceptible to measurement error by ART use. In 2011 the National Institute of Allergy and Infectious Diseases (NIAID) issued a call for studies of viral diversity measurement approaches [Reference Andersson20–Reference Laeyendecker23], followed in 2012 by a call for novel HIV incidence biomarker discovery projects from the BMGF [24]. These early-phase discovery projects (see Table 3) have resulted in a renewed emphasis on, and increased resources for, development, optimization and validation work.

Fig. 2. CEPHIA incidence assay critical path. Listed on the right are specific milestones that should be demonstrated before an assay moves forward to further development.
Table 3. Biomarker discovery and new assay development for HIV incidence measurement
BMGF, Bill & Melinda Gates Foundation; NIH, National Institutes of Health; CDC, Centers for Disease Control and Prevention; PBMC, peripheral blood mononuclear cell.
Repositories of blood and non-blood specimens, and construction and distribution of panels for incidence assay development and evaluation
Previously, a major barrier to development, optimization and independent evaluation of HIV incidence assays was the lack of availability of suitable specimens but the creation of relatively large specimen repositories for incidence assay evaluation by the Centers for Disease Control (CDC) and National Institutes of Health (NIH) has enabled rigorous assessment of several assays, leading to critical insights on the potential utility of RITAs [Reference Busch2, 4–Reference Duong6, Reference Laeyendecker23, Reference Mastro25–Reference Curtis28]. However, the specimens collected in these repositories were available in only limited volumes.
CEPHIA, on the other hand, received funding to build a larger repository that could facilitate direct comparative evaluations of assays and biomarker discovery. The effort focused on acquiring plasma specimens from subjects who had enrolled in prospective incidence cohorts from diverse countries of origin where the dates of detectable infection were known with a high level of confidence. Specimens were additionally acquired from collaborators who studied long-term HIV disease outcomes, response to antiretroviral treatment, and elite controllers, all of which are populations critical to the establishment of FRRs applicable to real-world settings. Plasma specimens with large volumes were sought, since the availability of multiple identical panels of pedigreed samples would greatly simplify the future task of comparative assay performance evaluations. Through active collaboration with existing clinical HIV research cohorts and blood banks interested in the development of HIV incidence assays, the CEPHIA repository now includes more than 9000 highly selected plasma specimens for which an estimated date of detectable infection could be calculated using a standardized approach, or which fell into specific other categories known to result in measurement error for recent HIV infection (elite controllers, ART-suppressed, etc.). Most samples in the repository have at least 10 ml of plasma available and they represent a broad diversity of subtypes and geographies (see Fig. 3). In the last 3 years, many of the Consortium's clinical collaborators have begun collecting alternative specimen types such as serum, dried blood spots, urine, saliva and stool, which have also been added to the CEPHIA repository to support evaluation and development of new tests and novel biomarkers. Relevant clinical and laboratory data, including detailed information on predicate HIV diagnostic testing, CD4+ T-cell counts, viral load, ART status and co-infections, are ascertained for all subjects/samples and maintained in a dedicated database. Where possible these specimens have been benchmarked against the currently best-performing incidence assays.

Fig. 3. Clade/geographical breakdown of CEPHIA repository specimens.
New specimen panels and support for biomarker discovery and assay development
CEPHIA specimen panels have been prepared and are available for biomarker discovery work or assay evaluation. Specimen information is accessible and searchable online, along with instructions for access, on the CEPHIA website (www.incidence-estimation.com/CEPHIA). Applications for repository access are reviewed to ensure that this valuable resource is used appropriately. Currently supported studies include both focused hypothesis-driven studies (for instance, how the gut inflammasome and specific subclasses of HIV antibodies change during the transition from recent to longstanding HIV infection); and non-hypothesis-driven efforts to identify signatures of recent HIV infection, including numerous types of novel biomarkers (e.g. searches for antibodies reactive to peptoids in a large ‘peptoid shape library’; exosomal or extracellular microRNAs; and urinary and blood metabolites). Table 3 summarizes a number of new approaches being undertaken. If successful, any of these new biomarkers will face the challenging transition from research assays to kit-based assays that can be successfully transferred and implemented in the field.
For assay developers, CEPHIA has developed relatively small panels ranging from 100 to 300 specimens in each panel. These panels are available in multiple specimen types and in multiple replicates.
-
• A recency biomarker screening panel containing 125 specimens from recent and longstanding infections, as well as HIV-negative specimens, is available for early stage explorations. This panel is available in a wide variety of specimen types.
-
• For more advanced biomarker candidates, a proof of concept panel can be distributed. To determine if a candidate marker has a plausible MDRI (e.g. 4–12 months), this panel contains 150 specimens with well-characterized EDDIs from the first 2 years post-infection, and 150 specimens from subjects known to have longstanding infections ranging from 2 to 11 years post-infection. These 300 specimens are balanced between B and C subtypes. There are an additional 50 ‘challenge’ specimens, which are 10 virally suppressed specimens from subjects treated within 60 days of the last plausible date of detectable infection, and 40 specimens treated later after infection but nonetheless virally suppressed.
Specimen panels for independently assessing performance of HIV incidence assays
For assays where preliminary data are available and an independent evaluation by the CEPHIA is desired, two specific specimen panels have been created:
-
• A qualification panel (n = 250) is provided under code (blinded) to researchers whose assays have reached an advanced and well-defined set of criteria, including that the assay is available in a kit format. The developers report blinded results that are evaluated by the CEPHIA group to confirm that the assay can reliably distinguish recent from longstanding infections.
-
• For assays that are thus qualified, an evaluation panel (n = 2500) is used by CEPHIA laboratories in blinded assay evaluations as part of a process described below. The composition of the evaluation panel allows determination of the effect of a number of known sources of measurement error for the performance of the assay (e.g. HIV subtype, antiretroviral treatment, elite controller status and advanced immunodeficiency) and enables determination of an MDRI and FRR for each assay (or combinations of assays). Each evaluation panel also includes multiple blinded aliquots of pedigreed samples (25 replicates of each) with antibody reactivity characteristic of recent, intermediate and longstanding infection to allow evaluation of the reproducibility of each candidate assay.
A process for independent evaluation of incidence assay performance
The development of TPPs, a specimen repository, and test panels that are now available represent a new standard for evaluations of HIV incidence assays. Independent evaluations that are undertaken should aim to match the CEPHIA principles of:
-
(1) Using comprehensive panels of specimens (with specimen background data blinded to the test operator) and appropriate specimens to ensure that statistical analyses of the results can quantify the impact of known sources of measurement error for incidence assays.
-
(2) Evaluations performed independently of the assay developers.
-
(3) Assay developers/manufacturers provide standard operating procedures, training, and certification of proficiency of the laboratory prior to initiating evaluations.
-
(4) Laboratories operate within a stringent quality system and with rigorous procedures in place to monitor and document the evaluations.
-
(5) All test output data analysed separately to the laboratory where results can be unblinded, verified for completeness, and compiled for analyses, ensuring all aspects of assay performance evaluated in addition to the qualitative output (recent/non-recent designations) from the assay.
To date, CEPHIA has completed 10 assay evaluations (see Table 4). Detailed analyses of individual assay performance have been presented at various meetings and conferences and individual assay performance summaries including assay usability are available on request (http://www.incidence-estimation.com/page/CEPHIA-assay-evaluations); a report summarizing data from the first five comparative assay evaluations has been published separately [Reference Kassanjee29].
Table 4. Main characteristics of assays previously evaluated by CEPHIA

DBS Dried blood spot; TBD, to be determined; GMP, good manufacturing practice.
Parameters chosen as representative of indicators described within target product profile (TPP). No differentiation of acceptability to TPP is given.
For consistency, FRR and MDRI calculated as per Kassanjee et al. [Reference Kassanjee29]. Figures used are manufacturer/developer-described parameters without changing thresholds to improve performance. MDRI and FRR calculated without use of a recent infection testing algorithm.
MDRI and FRR estimates not shown for Centers for Disease Control and Prevention (CDC) multiplex assay due to the multiple estimates available from the different combinations of analytes and conditions available within the single assay on which to base FRR and MDRI estimates.
These evaluations have highlighted the variability in assay performance and provided new insights on factors to be considered in using tests and designing RITAs. For instance, the effect of viral suppression on reliability of antibody-based incidence assays was shown to be highly dependent on the timing of ART initiation (i.e. FRRs are substantially higher in samples from persons treated early vs. later in infection). The analyses clearly indicate a need for inclusion of viral load results in interpretation of results from all antibody maturation-based incidence assays, to mitigate the impact of ART treatment and elite controllers on FRRs. Going forward, with the increasing uptake of ART and the availability of laboratory-based data on ART exposure to monitor progress towards the UNAIDS 90-90-90 targets, the inclusion of ART data in RITAs may become part of many national population-based survey protocols. In contrast, the same evaluation showed no evidence for a relationship between low CD4+ T-cell count and false-recent misclassification on the evaluated assays; this finding is important because it calls into question the need to perform CD4+ T-cell testing, which adds cost and logistical challenges in cross-sectional surveys. In another example, the benefit of independent evaluation, when compared to previous, developer-led studies, was highlighted when conflicting results between the evaluations contributed to the revision of published recommendations for interpretation of the Sedia™ HIV-1 LAg-Avidity assay [Reference Duong30].
A major obstacle now facing the incidence assay field is the cost and funding of future evaluations. As the funding from the BMGF comes to an end, identifying new support to maintain independent evaluation of future assays is critical. The cost of collecting suitable specimens, outside of existing systems, means any new evaluation may cost up to US$ 200 000 per evaluation, depending on requirements, which may be prohibitive to small companies trying to bring new products to market.
REMAINING CHALLENGES TO THE FIELD
Regulatory and policy issues
The potential use of assays to improve individual patient management and further prevention efforts through discriminating recently infected subjects following or coincident with initial HIV diagnosis could substantially increase the market for their use [Reference Busch2]. However, applying the tests to named patients samples with specific claims for use of results in clinical care will increase regulatory hurdles that might limit the application of assays in the field. To address these considerations, the CEPHIA evaluations were undertaken using a comprehensive quality assurance system to ensure that accurate and complete records of the independent evaluation are fully documented. All sample characteristics, test performance documentation, and results can be shared with regulatory bodies should a company seek a regulatory claim on a particular assay. The type and scope of information required for regulatory approval of recency assays has no precedent, and it remains to be seen whether the FDA, Council of Europe (CE) or other regulatory agencies will accept such evaluations as suitable for inclusion in a regulatory claim. Should this not be the case, then it is difficult to envisage where companies would be able to access sufficient numbers of appropriately characterized specimens to support an evaluation.
In many countries, pre- and post-market regulatory control for in-vitro diagnostics is often not adequate to ensure that assay use in their markets will meet international standards for safety, quality and performance. In these countries, national authorities may rely on the WHO for recommendations and advice. For assay developers, prequalification assessment by the WHO, alongside a performance evaluation by CEPHIA or similar group, may be an alternative to seeking regulatory approval through US FDA or CE marking, which is targeted for the use of assays in well-resourced laboratories rather than in resource-limited settings. WHO prequalification assessment consists of a review of a product dossier that substantiates the manufacturer's claims for safety, quality and performance alongside an inspection of the site(s) of manufacture to review the quality management system under which the assay is manufactured. The choice of pre-market assessment to be undertaken will depend on where the assay will be supplied, such as in settings of high HIV incidence and in settings with greatest need for HIV intervention studies.
Laboratory standards and external quality assurance
During the performance of the CEPHIA evaluations, laboratories often interpreted published methods differently. These subtle differences may affect the performance of the assay and lead to different results. This has highlighted the need for unambiguous standard operating procedures and for effective training in the performance and interpretation of assays, along with the need for an independent quality assessment system using blinded panels of well-characterized specimens to monitor assay performance across laboratories. Previously, such external quality assurance (EQA) programmes for the BED and Vironostika ‘detuned’ assays were supported by the US CDC but, as the use of these assays reduced, support for these programmes has been withdrawn. Because most incidence assays are either in-house (‘home-brew’) tests, modified versions of commercially available kits, or manufactured by small businesses with relatively limited QA resources, in-process kit controls are often unsuitable or insufficient to confirm that the assay is performing as expected in its modified format. Furthermore, it is unclear how sufficient funding could be obtained to support production and management of EQA panels, given the limited market for some assays.
In collaboration with CEPHIA and CDC, the External Quality Assurance Program Oversight Laboratory (EQAPOL), based at Duke University, has begun a proficiency testing programme for the LAg-Avidity assay. The success of this pilot effort offers hope that an independent EQA programme for incidence assays can be developed. However, some challenges remain in this area. The fact that a range of incidence assays are in use worldwide makes it challenging to predict which other assays need to be included long-term in the EQA scheme, and funding constraints to the EQAPOL approach may inhibit widespread participation of non-US-funded sites. Furthermore, there is an urgent need for EQA panels that include dried blood spot specimens as well as plasma.
Global coordination to advance development and application of incidence assays
The WHO Technical HIV Incidence Assay Working group (HIVIWG) has continued to meet on an annual basis (http://www.who.int/diagnostics_laboratory/links/hiv_incidence_assay/en/) to provide technical guidance and to advance efforts in this area of work (for example, see http://www.unaids.org/sites/default/files/media_asset/HIVincidenceassayssurveillancemonitoring_en.pdf). The WHO HIVIWG has supported a number of initiatives, especially related to training of staff in the use and interpretation of data and in the preparation of guidance documents. Other groups, including the HIV Modelling Consortium, UNAIDS Reference Group on Estimates, Modelling and Projections, and CEPHIA have met frequently during the 5 years since the 2011 Incidence Assay Critical Path meeting. However, it remains unclear how recommendations from groups such as CEPHIA will be implemented and endorsed by normative guidance agencies, or how the regulatory environment for incidence and recent infection assays will evolve in the post-CEPHIA era. There is uncertainty regarding funding for purchasing agreements that will guarantee assay supply on a continuing basis in the context of limited markets and reliance on small companies for the manufacture of many of these assays. The CDC's Global Health Initiative provides an example of one approach, but one that does not extend to all countries.
CONCLUSIONS
Over the last few years there has been a continued interest in the development and application of incidence assays, which have evolved from simple techniques involving the quality or quantity of antibody present to now include more diverse biomarkers. This will increase complexity as multiple and novel biomarkers are considered in determining recency, and will potentially lead to more specialized equipment being required for biomarker-based incidence estimation. The potential of molecular assays (e.g. viral sequence diversity) for use in HIV incidence estimates, which seemed considerable a few years ago, has not yet materialized. The use of incidence assays for studies other than cross-sectional incidence determination has increased, and the desire for the assays to be used on an individual patient basis is gaining momentum.
Further, rapidly changing national guidelines on the use of ART, resulting in earlier treatment of individuals and increasing use of pre- and post-exposure prophylaxis will render the interpretation of incidence assays more challenging in the future. This will require improvements in the current assays and a better understanding of how to interpret data within a changing and challenging environment.
The landscape for development of incidence assay approaches has also been changed. A more complete consensus has formed around a TPP and an assay development critical path. The formation of the CEPHIA has made more specimens, data, analysis tools and technical support available to researchers than ever before. However, the lack of coordinated action on purchasing agreements between funders, governments and developers will likely slow the roll-out of these assays, and uncertainty remains about assay regulation and provision of external quality assurance. Given the considerable funds being invested in national surveys, it is disturbing that currently there is only very limited funding available for proper quality control, training and evaluation of HIV incidence assays. The development of sustainable funding for this area is critical to ensure the quality and accuracy of results, if recent and future developments are to be translated into meaningful public health interventions.
APPENDIX. CEPHIA Collaborators
The Consortium for the Evaluation and Performance of HIV Incidence Assays (CEPHIA) is comprised of the authors and: Tom Quinn, Oliver Laeyendecker (Johns Hopkins University); David Burns (National Institutes of Health); Anita Sands (World Health Organization); Tim Hallett (Imperial College London); Sherry Michele Owen, Bharat Parekh, Connie Sexton (Centers for Disease Control and Prevention); Anatoli Kamali (International AIDS Vaccine Initiative); David Matten, Hilmarié Brand, Trust Chibawara (South African Centre for Epidemiological Modelling and Analysis); Elaine Mckinney, Jake Hall (Public Health England); Mila Lebedeva, Dylan Hampton (Blood Systems Research Institute); Lisa Loeb (The Options Study – University of California, San Francisco); Steven G. Deeks, Rebecca Hoh (The SCOPE Study – University of California, San Francisco); Zelinda Bartolomei, Natalia Cerqueira (The AMPLIAR Cohort – University of São Paulo); Breno Santos, Kellin Zabtoski, Rita de Cassia Alves Lira (The AMPLIAR Cohort – Grupo Hospital Conceição); Rosa Dea Sperhacke, Leonardo R. Motta, Machline Paganella (The AMPLIAR Cohort – Universidade Caxias Do Sul); Helena Tomiyama, Claudia Tomiyama, Priscilla Costa, Maria A. Nunes, Gisele Reis, Mariana M. Sauer, Natalia Cerqueira, Zelinda Nakagawa, Lilian Ferrari, Ana P. Amaral, Karine Milani (The São Paulo Cohort – University of São Paulo, Brazil); Salim S. Abdool Karim, Quarraisha Abdool Karim, Thumbi Ndungu, Nigel Garret, Nelisile Majola, Natasha Samsunder (CAPRISA, University of Kwazulu-Natal); Denise Naniche (The GAMA Study – Barcelona Centre for International Health Research); Inácio Mandomando, Eusebio V Macete (The GAMA Study – Fundacao Manhica); Jorge Sanchez, Javier Lama [SABES Cohort – Asociación Civil Impacta Salud y Educación (IMPACTA)]; Ann Duerr (The Fred Hutchinson Cancer Research Center); Maria R. Capobianchi (National Institute for Infectious Diseases ‘L. Spallanzani’, Rome); Barbara Suligoi (Istituto Superiore di Sanità, Rome); Susan Stramer (American Red Cross); Phillip Williamson (Creative Testing Solutions/Blood Systems Research Institute); Marion Vermeulen (South African National Blood Service); and Ester Sabino (Hemocentro do Sao Paolo).
DECLARATION OF INTEREST
None.







