


INTRODUCTION
Staphylococcus aureus is recognized as one of the most prevalent pathogens isolated from hospitalized patients and is of increasing importance in the community setting [Reference Boyle-Vavra and Daum1]. It is associated with a wide spectrum of infections ranging from mild superficial skin lesions to life-threatening systemic infections [Reference Ma2, Reference Johnsson3]. The ongoing evolution of antimicrobial resistance particularly methicillin-resistant strains of S. aureus (MRSA) has complicated the treatment of infections with such isolates. Hospital-acquired MRSA (HA-MRSA) strains have been and remain endemic in many countries worldwide, but in recent years community-acquired (CA-MRSA) strains have emerged to cause serious invasive and life-threatening infections in young, healthy patients without significant healthcare contacts [Reference Boyle-Vavra and Daum1].
Resistance to methicillin is encoded by the staphylococcal cassette chromosome mec (SCCmec) element, composed of the mec gene complex, and the ccr (cassette chromosome recombinase) gene complex, encoding for the recombinase gene [Reference Ma2, Reference Ito4, Reference Ito5]. SCCmec elements have been classified into eight major types (I–VIII) some of which are differentiated further into subtypes [Reference Chambers and DeLeo6]. SCCmec types I, II, and III, and types IV and V have been associated with HA- and CA-MRSA, respectively [Reference Ito4, Reference Ito5, Reference Qi7]. CA-MRSA lineages have also been identified in healthcare-associated infections, sometimes causing invasive bloodstream infections [Reference Trindade8, Reference Popovich9].
The production of Panton–Valentine leukocidin (PVL) is highly associated with the most common genetic lineages of CA-MRSA particularly those prevalent in North America [Reference Deurenberg and Stobberingh10, Reference Vandenesch11] but some lineages identified in the Far East lack the toxin [Reference Yamamoto12, Reference Hoa13]. Although PVL has been shown to be a significant virulence factor in necrotizing pneumonia [Reference Labandeira-Rey14, Reference Gillet15], its role in promoting the virulence of CA-MRSA in skin and soft tissue infections has been questioned [Reference Voyich16].
The most widely used methods for genotyping S. aureus are multi-locus sequence typing (MLST) and pulsed-field gel electrophoresis (PFGE) [Reference Aires de Sousa and de Lencastre17]. MLST groups strains into sequence types (STs), and these can be sorted by their relatedness into clonal complexes (CCs) [Reference Deurenberg and Stobberingh10, Reference Grundmann18]. Enright et al. identified 11 major MRSA clones within five groups of related genotypes in a large international strain collection of S. aureus and suggested that major MRSA clones evolved from successful epidemic methicillin-sensitive strains [Reference Enright19]. DNA macrorestriction analysis by PFGE on the other hand, is excellent for the determination of strain relatedness in outbreak situations but does not inform the evolutionary relatedness of the overall populations of strains [Reference Grundmann18]. It also lacks the portability of MLST owing to difficulties in reproducibility between laboratories [Reference Deurenberg and Stobberingh10]. Genetic analysis of strain types of S. aureus can also be performed by spa-sequence typing. A region of the spa gene consists of a variable number of direct repeats which exhibit extensive strain-dependent polymorphisms [Reference Brigido20]. Its advantage over the preceding methods is that it provides data informative of the population structure and evolution of strains as well as identifying strain relationships for studies of hospital outbreaks. However, the discriminating power of the method is sometimes limited by the occurrence of the same or related spa types in different clonal lineages [Reference Deurenberg and Stobberingh10].
There have been several studies on the molecular characterization of S. aureus isolates in the Middle East, two of which revealed the prevalence of ST239 MRSA isolates in Tehran and Turkey [Reference Fatholahzadeh21, Reference Alp22]. The absence of such reports from Lebanon provided the rationale of the present study of the molecular characterization of S. aureus isolated from Lebanon.
METHODS
Bacterial isolates
A total of 130 S. aureus clinical isolates recovered from different patients between January 2006 and May 2007 at the Clinical Microbiology Section of the American University of Beirut in Lebanon was used in this study. The isolates were randomly chosen targeting primarily those recovered from wound and respiratory specimens. DNA was extracted using QIAamp DNA Mini extraction kit (Qiagen, USA) and the efficiency of the DNA extraction method was tested using 16S rDNA PCR [Reference Tokajian23].
Multiplex PCR for SCCmec subtyping
The SCCmec Multiplex-PCR (M-PCR) assay for types and subtypes I, II, III, IVa, IVb, IVc, IVd and V and PCRs for mecA and ccr genes were as previously described [Reference Zhang24]. The following control strains for SCCmec types and subtypes were kindly provided by Dr T. Ito; NCTC 10442 (type I), N315 (type II), 85/2082 (type III), JCSC 4744 (type IVa), JCSC 2172 (type IVb), JCSC 4788 (type IVc) and WIS (type V). All PCR assays were performed using 1·0 U platinum Taq DNA polymerase (Invitrogen Inc., USA). The SCCmec subtype IVc failed to amplify even with the control strain and so primers for this subtype were replaced by the set recommended by Kondo et al. [Reference Kondo25]. All PCR assays run incorporated a negative control (without template DNA), and a PCR control with Escherichia coli DNA. PCR detection of the PVL gene was performed as previously described [Reference Lina26] with S. aureus ATCC 49775 as a positive control.
spa typing
The polymorphic X region of the protein A gene (spa) was amplified using the primers spa-1113f and spa-1514r [Reference Harmsen27]. All sequencing reactions were carried out using the ABI Prism BigDye Terminator version 3.1 cycle sequencing ready reaction kit (Applied Biosystems, USA). spa types were assigned using the Ridom StaphType software version 1.5.13 (Ridom GmbH, Germany) as described by Harmsen et al. [Reference Harmsen27] and further grouped into spa clonal complexes (spa CCs) using the BURP algorithm with the calculated cost between members of a group being ⩽4 and excluding spa types fewer than five repeats [Reference Strommenger28].
MLST
MLST was performed on 15 isolates of the most common spa types by sequencing the arc, aroE, glp, gmk, pta, tpi and yqiL genes as described by Enright et al. [Reference Enright19] using the BigDye Terminator version 3.1 cycle sequencing ready reaction kit (Applied Biosystems). MLST types were assigned by submitting the sequences to the S. aureus database on the website (http://www.mlst.net/).
RESULTS
Table 1 shows that approximately equal numbers of S. aureus isolates were recovered from outpatients and in-patients. The majority (93/130) were MRSA. More than half of the MRSA and a third of the methicillin-sensitive S. aureus (MSSA) isolates were from cutaneous sites (wounds, cysts, abscesses). The prevalence of the PVL gene in MRSA isolates was 62% compared to 20% in MSSA isolates (Table 1). Most (87%) of MRSA isolates were of SCCmec type IVc followed by type III (10%), with types II, IVb and V occurring in 1% each. The PVL gene was detected only in isolates harbouring SCCmec type IVc, and in the single isolate with type IVb.
Table 1. Demographics and molecular characterization of MRSA and MSSA isolates from Lebanon
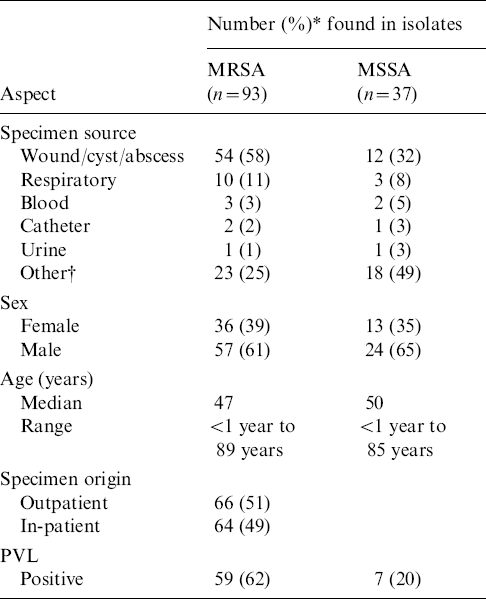
PVL, Panton–Valentine leukocidin.
* Decimals are rounded.
† Other: sputum, DTA, brain, neck mucus, urine, gall bladder, joint fluid, semen, tissue.
spa typing
All isolates were assigned to 48 different spa types, varying in length between 2 (t524) and 16 (t032) repeats. The most common spa types identified were t044 (37·7%) and t037 (4·6%), and occurred only in MRSA isolates. All spa t044 isolates harboured SCCmec type IVc and the PVL gene, except for isolate S44, which was of type III and PVL negative. It is noteworthy that a novel repeat was identified in this study which resulted in a new type designated as spa t4099; this isolate was PVL positive with SCCmec subtype IVc.
Using the BURP algorithm, spa types were clustered into 23 different groups, with 11 groups comprising more than one spa type and 12 ‘singletons’. Since clustering parameters excluded spa types shorter than five repeats, one type (t524) was excluded. Six of the 11 spa CCs had designated group founders (spa CC021, spa CC008, spa CC660, spa CC044, spa CC279, spa CC005). The group founder, if present in a group, is defined as the spa type with the highest founder score and is assigned to the spa type with the most closely related spa types and strains. Twenty-eight different spa types were identified in the MSSA isolates compared to 27 in the MRSA isolates. Both groups of isolates were distributed into all identified clusters, except for spa CC44, which comprised only MRSA strains. CC 21 (spa CC21) and CC44 together accounted for 60% of the isolates; 16% of MSSA isolates fell in CC5 and CC8.
MLST
MLST revealed 11 STs in the 15 isolates tested. There were eight major MRSA clones defined as isolates with the same ST and the same SCCmec type. These clones were associated with complexes CC80, CC30, CC8, CC22, CC5, CC97 and CC6 (Table 2). ST80, PVL-positive, SCCmec subtype IVc, was the predominant genotype in the 15 tested population. PVL gene positive MRSA fell in clones CC80, CC30 and CC5 and PVL-negative isolates in other clonal complexes. None of the PVL-positive MRSA had the same clonal class as the PVL-negative MRSA. However, MSSA strains were distributed within CC5, CC30, CC80, CC1, CC5 and CC121.
Table 2. An overview of the major MRSA and MSSA spa types and their corresponding MLST clones recovered from Lebanon

ST, MLST sequence type; CC, clonal complex; sg, singleton.
DISCUSSION
The present study is, to our knowledge, the first comprehensive comparison of the genetic background of isolates of MRSA and MSSA in Lebanon. It revealed that the majority of MRSA harboured the PVL gene and the most prevalent clone was ST80-MRSA-IVc, spa type 44. Three out of the five major PVL-positive CA-MRSA clones (ST80, ST30, ST8) disseminating worldwide were detected in this collection, and the identification of ST5-MRSA-IVc with the PVL gene was of particular concern since this lineage has been associated with a high capacity to spread and thus become epidemic [Reference Deurenberg and Stobberingh10]. The distribution of isolates with the predominant SCCmec subtype IVc (86%) according to specimen source revealed that almost 75% were from soft tissue and 20% from respiratory sources, which is in accord with several other studies [Reference Faria29] as was the distribution of SCCmec elements and the frequency of PVL gene positivity in different age groups, although some reports have highlighted PVL-positive MRSA mainly in younger people [Reference Millar30–Reference Huang32].
A variety of genotyping techniques is available to determine staphylococcal clonal relatedness. Strommenger et al. [Reference Strommenger33] have previously shown a wide similarity of clustering results obtained by spa typing/BURP analysis with those obtained by well-established methods (SmaI macrorestriction analysis and MLST/eBURST) while Enright et al. used ST and SCCmec type to define MRSA clones [Reference Enright19]. However, a potential problem with spa typing is that it involves sequencing of only one small region of the chromosome, which is subject to recombination between unrelated clones. This could result in isolates exhibiting the same spa type when they are unrelated by other methods [Reference Cookson34]; spa t037 isolates have arisen from a single recombination event that involved the exchange of a 200-kb DNA fragment including the spa gene between MLST30 and MLST239 [Reference Robinson and Enright35, Reference Holtfreter36].
MSSA strains in this study were distributed within CC5, CC30, CC80, CC1, CC5 and CC121; the majority of which represent international MSSA pandemic clones [Reference Strommenger33]. The increased diversity within the MSSA isolates was in accord with the findings of Strommenger et al. [Reference Strommenger28].
Of the eight major MRSA clones (ST80, ST30, ST8, ST239, ST22, ST5, ST97, ST6) identified here, the ST8 clone is known to have disseminated in Europe and USA, ST30 in Australia, Europe and South America, ST80 in Asia, Europe and the Middle-East, ST5 in Africa, Europe and South America and ST22 in parts of Europe. Other MRSA clones detected in this study included: ST30-MRSA-IVc observed in Australia, Europe and South America, ST1-MRSA-IVc clone observed in Asia, Europe and USA and ST239-MRSA-III the Brazilian/Hungarian clone [Reference Robinson and Enright35].
ST80-MRSA-IVc, the major MRSA clone in Lebanon, was frequently associated with skin sources (wounds, abscesses) which is in accord with Larsen and colleagues who found that the ‘European CA-MRSA’ clone CC80-MRSA-IV primarily caused CA infections, predominantly skin and soft tissue, in young people outside hospitals [Reference Larsen37], with some of the isolates being recovered from hospitalized patients. However, European CA-MRSA seemed less adapted to persist in hospital environments, and patients with a recent history of travel or family relation to the Mediterranean or Middle East were highly overrepresented in their study.
In conclusion, we present here the first comprehensive study on the genetic population structure of S. aureus clinical isolates from Lebanon. The widespread distribution of the PVL gene positive ST80-MRSA-IV clone in different regions of the world is probably related to international travel. If the prevalence of this clone reflects increased community acquisition of S. aureus infections in Lebanon, focused studies are urgently needed to define the disease burden and to establish their association with clinical presentation and patient outcomes. The data presented here represent a starting point defining the major genetic populations of both MRSA and MSSA in Lebanon and provide a basis for clinical epidemiological studies to determine their prevalence in disease and carriage and inform the development of measures to control the spread of these potentially serious infections.
ACKNOWLEDGEMENTS
This research was partially supported by funds from Orfaleae Family Foundation and the Lebanese American University Research Council.
DECLARATION OF INTEREST
None.


