


INTRODUCTION
Helicobacter pylori (H. pylori) is a common pathogen associated with human gastric and extragastric diseases. The failure of the host immune response to clear this infection results in chronicity. H. pylori infection is associated with the gastro-duodenal diseases that include chronic gastritis, peptic ulcer, i.e. gastric and duodenal ulcer, gastric carcinoma (GC) and mucosa-associated lymphoid tissue lymphoma (MALToma) [Reference Wroblewski, Peek and Wilson1]. H. pylori infection elicits an acute-phase response that activates an immune host response causing an imbalance between cell proliferation and cell apoptosis [Reference Nurgalieva2]. The gastric colonization of H. pylori depends on several factors that include urease enzyme, cytotoxin-associated geneA (cagA), vacuolating cytotoxin A (vacA) and H. pylori outer membrane proteins [Reference Kao, Sheu and Wu3]. The genetic diversity among H. pylori strains accounts for varying manifestations among persons colonized with H. pylori [Reference Hopkins, Girardi and Turney4]. The virulence of H. pylori is determined by markers, e.g. the cytotoxin-associated gene pathogenicity island (cag-PAI), vacA alleles, outer immunoproteinA and membrane protein Q (HopQ) type [Reference Yakoob5–Reference Achtman9].
The genetic diversity in H. pylori is associated with high mutation rate [Reference Morelli10, Reference Kennemann11]. Infection with multiple strains promotes recombination between them. Genes encoding H. pylori outer membrane proteins are significant among the imported DNA fragments [Reference Morelli10]. H. pylori genome sequences possess a large family of Hop genes [Reference Ilver12]. H. pylori strains expressing HopQ have facilitated attachment to gastric epithelial cells [Reference Loh13]. Previously, we studied 241 H. pylori strains that showed HopQ type 1 in 70 (29%), type 2 in 60 (25%), and types 1 and 2 in 111 (46%) strains, respectively [Reference Yakoob5].
H. pylori infection predates the development of gastric lymphoma [Reference Wotherspoon6]. Previously, an association between H. pylori and gastric low-grade B-cell non-Hodgkin lymphoma (B-cell NHL) was described [Reference Lehours14]. H. pylori was less common in high-grade gastric B-cell NHL in 38–51% with no specific mucosa-associated lymphoid tumor (MALT) features [Reference Wotherspoon6, Reference Lehours14]. H. pylori infection causes a chronic immuno-inflammatory reaction. This antigenic stimulation leads to lymphoid hyperplasia and acquisition of genetic instability, which follows activation of intracellular pathways [Reference Pereira and Medeiros15]. The disease progresses with cellular proliferation, resistance to apoptosis and emergence of a malignant clone [Reference Pereira and Medeiros15].
Ours is a low-risk region for GC with a high prevalence of H. pylori infection [Reference Yakoob16]. In a retrospective study that looked at the GC over a period of 10 years, 42 (11%) were cases of B-cell NHL [Reference Yakoob16]. In the current study, we determined the distribution of H. pylori and its virulence marker, i.e. cagA, cagAP, cagE, cagT, LEC, vacA alleles, and HopQ types 1 and 2 in B-cell NHL and compared their distribution with H. pylori strains associated with non-ulcer dyspepsia (NUD) and chronic gastritis.
MATERIAL AND METHODS
Patients
One hundred and seventy patients with upper gastrointestinal symptom that included abdominal pain were enrolled from the endoscopy unit extending from November 2012 to June 2016. Their mean age was 48 ± 13 years and range 21–83, male:female ratio was 1·4:1. Ours is a tertiary care center where the healthcare facilities are being availed by patients from all over the country. The hospital is located in a cosmopolitan city with an urban population of over 16 618 million in 2015. Population has varying ethnicity contributed by all the provinces of the country. Socio-economic status (SES) of majority of our patients varies from low to middle SES. One hundred and fourteen (67%) were diagnosed as NUD and in 56 (33%) B-cell NHLs. Other NHL subtypes, e.g. follicular, mantle, etc. were not found in this cohort of patients. Institutional ethics committee approved the study. Informed consent was obtained from all patients. The patients enrolled were not on any medications, such as antibiotics, H2-receptor antagonists, proton pump inhibitors, bismuth compounds in the last 3 months. A note was made of presenting symptoms and endoscopic findings (Table 1). Two gastric biopsies for histology were collected in formalin and four in normal saline two each for H. pylori culture and polymerase chain reaction (PCR). The PCR for HopQ alleles identified in H. pylori were single (i.e. type 1 or type 2) or multiple types (i.e. type 1 and type 2). CagA, cagA-promoter region (cagAP), cagE, cagT, left end of the cagA gene (LEC) and vacA alleles, i.e. s1a, s1b, m1, m2 and s2 were analyzed. Patients with H. pylori infection were treated with Bismuth-based quadruple therapy, while patients diagnosed with B-cell NHL were referred to oncologist for further management.
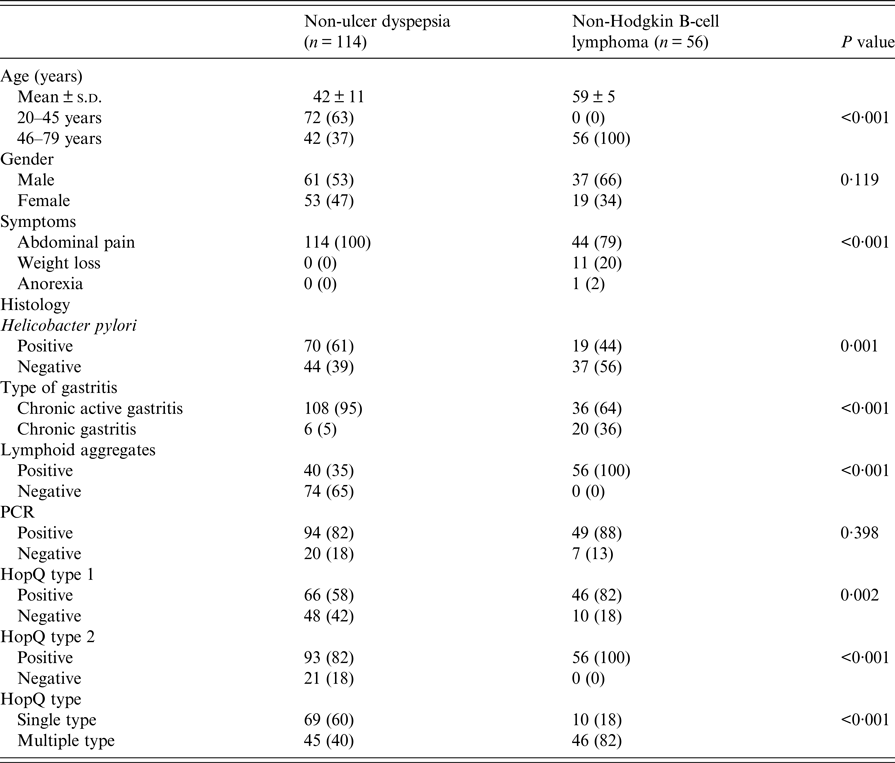
Table 1. Comparison of patients with non-ulcer dyspepsia and non-Hodgkin B cell lymphoma
H. pylori culture
Each specimen was homogenized in eppendorf tubes with electric homogenizer and inoculated onto Columbia blood agar (Oxoid) medium supplemented with Dents supplement (containing trimethoprim, vancomycin, amphotericin B and cefsulodin) and 7% defibrinated sheep blood and incubated at 37 °C under microaerophilic conditions using anaerobic jars and strips (Campygen strips, Oxoid, UK) for 5 days producing microaerophilic conditions essential for the growth. Plates were then examined for bacterial growth. The identity of H. pylori was confirmed by colony morphology, Gram stain and production of urease and catalase. H. pylori isolates were defined as Gram-negative spiral-shaped bacilli that were urease and catalase positive.
Histology
An expert pathologist, unaware of the clinical details, reviewed all the biopsies and reported the diagnosis. All cases were shown to be of B-cell phenotype by immunochemistry with a panel of antibodies that included CD20, CD19, CD79a, CD22 and CD3. Diagnosis of B-cell NHLs was based on a positive CD20, CD19, CD79a and CD22· [Reference Nakamura, Müller-Hermelink, Bosman, Carneiro, Hruban and Theise17]. The criteria used for the diagnosis of B-cell NHL included diffuse sheets of large, blastic lymphoid cells, two to four times larger than normal lymphocytes, often infiltrating and destroying the gastric glandular architecture. Criteria for diagnosis of low-grade lymphoma of MALT type were as defined consisting of a diffuse proliferation of cells with epithelium infiltration forming characteristic lymphoepithelial lesions [Reference Isaacson18]. High-grade tumors were diffuse infiltrates of large blast cells [Reference de Jong19]. Following endoscopy patients underwent whole-body computerized tomographic scan to determine the extent of involvement. Patients were referred to oncology service for further management. There were 15 cases of low-grade and 41 of high-grade B-cell NHL. Paraffin-coated gastric tissues were stained with hematoxylin and eosin to study histopathology and H. pylori [Reference Price20].
Extraction of genomic DNA
DNA was extracted from gastric biopsy tissue as previously described [Reference Van Zwet21].
PCR
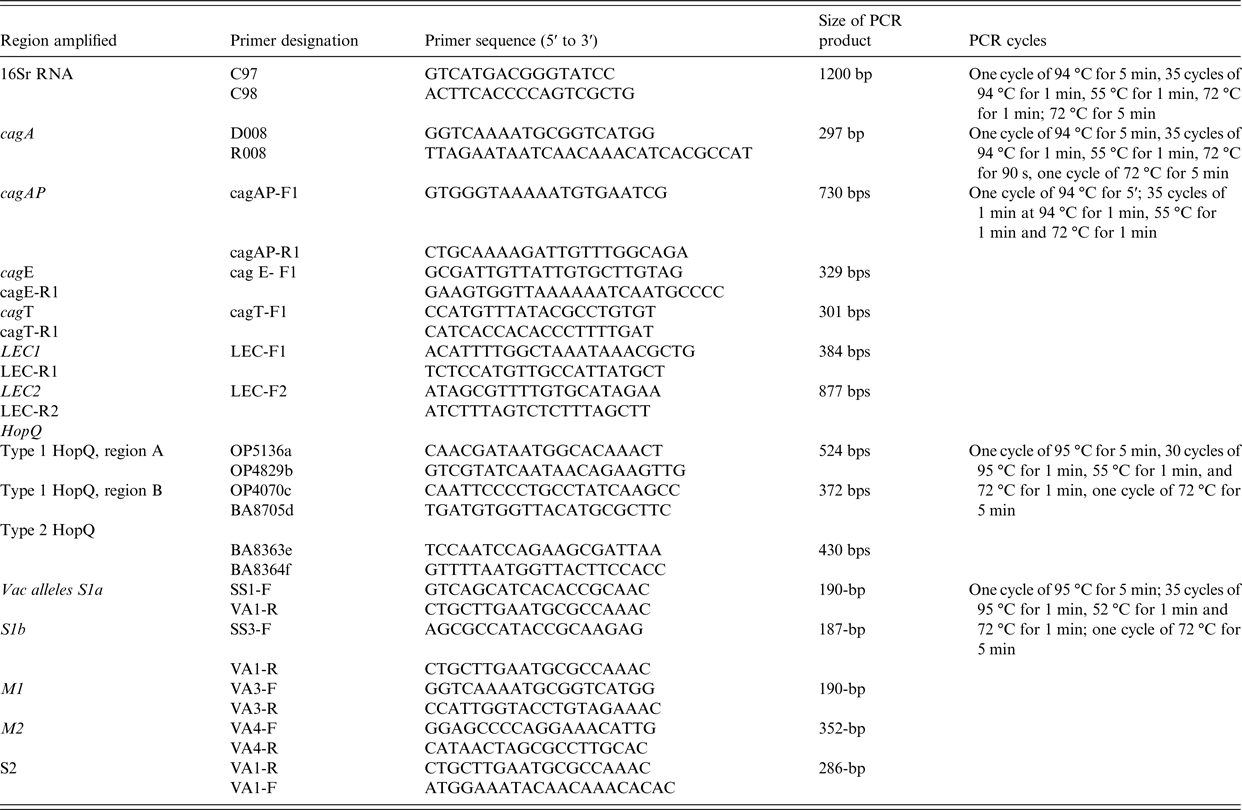
Amplification of 16S rRNA, cagA, cagAP, cagE, cag LEC and cagT and vacA alleles by PCR was performed by the method previously used before [Reference Yakoob22] (Table 2).
Table 2. Primers used in PCR experiments
HopQ genotyping
The HopQ type 1 and type 2 were determined by PCR methods described previously [Reference Cao and Cover23] (Table 2).
Sequencing of PCR product and BLAST Query
Sequence analysis was carried out by Macrogen (Seoul, South Korea). HopQ type 1 and type 2 sequences were published in our previous study and deposited in GenBank with accession numbers (KJ946296-KJ946308) and (KJ946309-KJ946314), respectively [Reference Yakoob5].
Sample size
The study determined the distribution of HopQ types in patients with B-cell NHL. From previous studies, H. pylori was present in 46% B-cell NHL [Reference Ferreri and Montalbán24] HopQ type 1 in GC in 53% and type 2 isolates in NUD in 41% [Reference Yakoob5]. Gastric ulcers were associated with H. pylori infection with multiple HopQ alleles in 46% compared with 23% with HopQ type 1. Therefore, the frequency of 46% with 95% level of confidence and (0·074) 7% bound on the error of estimation, a sample size of 175 patients was required. A total of 134 patients were required to establish their association with gastritis with 80% power. A sample size of 175 patients was required to cover both the study objectives.
Statistical analysis
Pearson χ 2, Fisher exact or likelihood ratio test were used where appropriate. A P value of <0·05 was significant. Data were analyzed using the SPSS version 19.0.
RESULTS
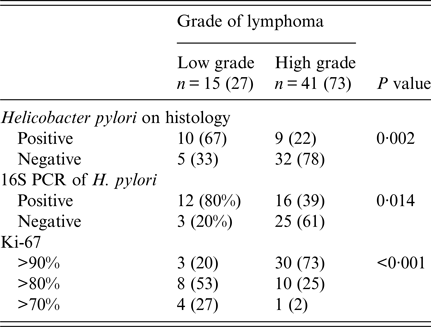
Patients with NUD were significantly younger than with B-cell NHL (Table 1). Abdominal pain was also significantly common in patients with NUD (Table 1). Endoscopically, there was gastritis in 114 (100%) NUD patients, while in B-cell NHL, there was a mass lesion 51 (91%) (P < 0·001). On histology, H. pylori was positive in 70 (61%) with NUD compared with 19 (34%) (P = 0·001) with B-cell NHL (Table 1). Lymphoid aggregates were common in B-cell NHL 56 (100%) compared with 40 (35%) (P < 0·001) in NUD (Table 1). Majority of B-cell NHL were diffuse large B cell that were high grade 41 (73%) with 30 out of 41 having >90% proliferative index (Table 3).
Table 3. Distribution of non-Hodgkin B-cell lymphoma
n (%), number and percentage.
HopQ allelic type
HopQ type 1 was positive in 66 (58%) in NUD compared with 46 (82%) (P = 0·002) in B-cell NHL; while HopQ type 2 was positive in 93 (82%) with NUD compared with 56 (100%) (P < 0·001) in B-cell NHL (Table 1). Single HopQ allele was present in 69 (60%) with NUD compared with 10 (18%) in B-cell NHL, while multiple HopQ was present in 46 (40%) in NUD compared to 46 (82%) (P < 0·001) in B-cell NHL (Table 1).
Cag-PAI gene
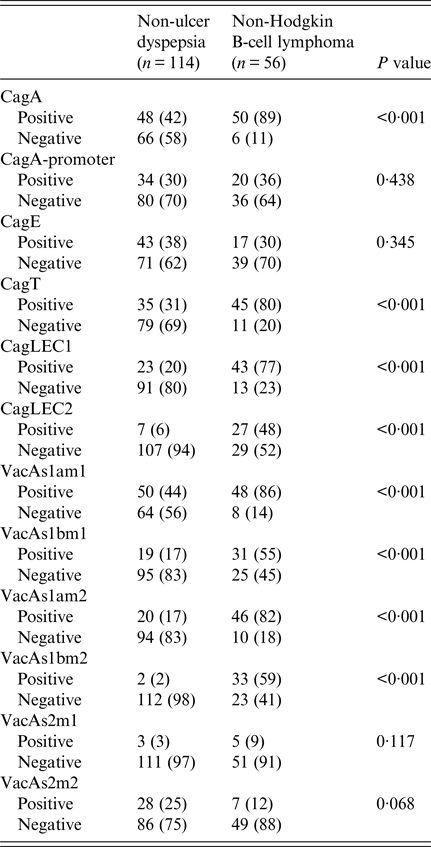
CagA was positive in 48 (42%) in NUD compared with 50 (89%) (P < 0·001) in B-cell NHL; cagT was positive in 35 (31%) in NUD compared with 45 (80%) (P < 0·001) in B-cell NHL; cag LEC1 was positive in 23 (20%) in NUD compared with 43 (77%) (P < 0·001) in B-cell NHL; while cag LEC2 was positive in 7 (6%) in NUD compared with 27 (48%) (P < 0·001) in B-cell NHL (Table 4).
Table 4. Comparison of Helicobacter pylori virulence groups marker in different
n (%), number and percentage.
VacA alleles
VacAs1am1 was positive in B-cell NHL in 48 (86%) (P < 0·001) compared with 50 (44%) in NUD, while s1bm1 was positive in 31 (55%) (P < 0·001) in B-cell NHL compared with 19 (17%) in NUD; s1am2 was positive in 20 (17%) in NUD compared with 46 (82%) (P < 0·001) in B-cell NHL; while s1bm2 was positive in 2 (2%) in NUD and in 33 (59%) (P < 0·001) in B-cell NHL (Table 4).
DISCUSSION
PCR for the H. pylori 16S rRNA PCR did not demonstrate any difference in the association of H. pylori with NUD and B-cell NHL (Table 3). This is particularly likely if H. pylori tested on the biopsy obtained from the B-cell NHL involved area, which may be necrotic and has low H. pylori load. PCR for H. pylori is known to have a higher yield [Reference Yakoob25]. Chronic active gastritis was predominant in both groups (Table 1). Infiltration of the gastric mucosa with neutrophils and lymphocytes leads to increased apoptosis and clearance of epithelial cell. H. pylori-induced macrophages and dendritic cells (DCs) that are known to secrete tumor necrosis factor-α that further promote inflammation [Reference Fan26]. After activation via Toll-like receptors, DCs are known to activate T cells to Th1 or Th2 response by expression of interleukin (IL)-12 or IL-10, respectively [Reference Bland27]. Dendritic cells exposed to H. pylori for 48 h exhibited a markedly attenuated ability to induce interferon-γ production that contributed to the persistence of the infection [Reference Hafsi28].
HopQ has a role in the pathogenesis of gastric B-cell NHL as it promotes attachment of the H. pylori to the epithelial cells. H. pylori binds to host cell receptors via adhesion molecule of the carcinoembryonic antigen (CEACAM) located on the gastric epithelial cell membrane to translocate cagA into host cells [Reference Javaheri29]. Multiple types of HopQ types were associated with B-cell NHL compared with the NUD (Table 1). In patients with B-cell NHL, both HopQ type 1 and HopQ type 2 were present and a multiplicity of the HopQ types was demonstrated in 82% of B-cell NHL (Table 1). In an animal study, naïve B cell exposed to H. pylori demonstrated a biphasic response in which low multiplicity of infection (MOI) (1–10) induced cellular proliferation and markedly inhibited apoptosis [Reference Bussiere30, Reference Delchier31]. Low levels of H. pylori infection that occur in vivo are associated with B-cell survival and proliferation, consistent with their potential to evolve into MALToma. The difference in the clinical outcome was not only due to the longer duration of the H. pylori infection in the B-cell NHL but was also contributed to by the virulence of the infecting H. pylori strains.
The limitation of this study is that we did not do immunohistochemical (IHC) staining to detect the genetic aberrations such as t(11;18)(q21;q21) and t(1;14)(p22;q32) that are associated with H. pylori-independent B-cell NHL [Reference Liu32]. For patients without t(11;18)(q21;q21) or t(1;14)(p22;q32), nuclear translocation of BCL10 and nuclear factor-κB (NF-κB) detected by IHC is predictive of H. pylori-independent state [Reference Yeh33]. CagA expression in tumor cells, particularly with nuclear expression is a useful biomarker in lymphoma cells, and is associated with the direct lymphomagenic effect of H. pylori on B cells. The titers of anti-H. pylori and anti-cagA antibodies were not checked. These titers have been reported to be significantly higher in H. pylori-dependent cases than in H. pylori-independent cases of t(11;18)(q21;q21)-negative gastric MALT lymphoma [Reference Peng34, Reference Eck35]. In the presence of cagA, B-cell lymphocytes evade apoptosis through the inhibition of p53 accumulation [Reference Sumida36, Reference Umehara37]. An important limitation of this study is that lymphoma patients are older than non-lymphoma patients, raising the possibility that lymphoma is a consequence of duration of infection, age, or some other factor, apart from H. pylori pathogenicity factors. The high mutation rates of H. pylori and chronic infection in the lymphoma patients may involve strains lacking the pathogenic markers. The cross-sectional nature of this study cannot account for the duration of infection with more or less pathogenic strains. However, these limitations do not invalidate the study.
H. pylori cagA was significantly associated with B-cell NHL (Table 4). This is consistent with a previously reported cagA 78–100% association with B-cell NHL [Reference Delchier31]. CagPAI genes, i.e. cagA promoter and cagE genes, were not significantly associated with either of the two conditions (Table 4). This variability may be attributed to DNA motifs that exhibited sequence heterogeneity in the cagA gene [Reference Loh38]. The cagPAI in B-cell NHL appears to be partially truncated as cagA promoter was 20 (36%) and cagE 17 (30%), respectively, compared with cagT and cagA LEC (Table 4). In an earlier local study, the presence of the cagA did not signify an intact cagPAI [Reference Yakoob39]. Most of the H. pylori strains studied had partial cagPAI with missing cagE and cagAPs [Reference Yakoob39]. CagA-positive H. pylori strains are potent in induction of host inflammatory responses, including activation of neutrophils, which releases highly genotoxic oxygen reactive species that induces barrier dysfunction and apoptosis in the gastric epithelium. CagA activation of the NF-κB affects cell proliferation through c-Fos and c-Jun and impaired immune response by inducing apoptosis of T cells [Reference Meyer-ter-Vehn40]. In our study, cagT and LEC genes were significantly associated with B-cell NHL (Table 4). CagE is required for the induction of IL-8 by host cells [Reference Owen41] and is a component of the H. pylori Type 4 Secretion System (T4SS) [Reference Fronzes, Christie and Waksman42]. It was also equally common in the H. pylori strains associated with NUD and B-cell NHL (Table 4). The cagT gene encoded an extracellular lipoprotein of the T4SS complex that stabilizes the other proteins [Reference Fronzes, Christie and Waksman42]. H. pylori also induces IL-8 via T4SS constituent cagL interaction with the host receptor integrin b1 and the subsequent activation of the mitogen-activated protein kinases and NF-kB pathway [Reference Gorrell43].
VacA s1 alleles were significantly associated with B-cell NHL compared with NUD (Table 4). Both vacAs1/m1 and s1/m2 are virulent form common in gastric diseases [Reference Fischer44]. VacAs1/m2 has been variably reported about its association with MALToma [Reference Lehours14, Reference Koehler45]. The underlying mechanisms involve v acA blocking antigen presentation to T cells [Reference Torres46], T-lymphocyte activation [Reference Boncristiano47] and maturation of macrophage phagosomes [Reference Sundrud48], thus suppressing T-cell responses to H. pylori and contributing to the immunosuppression and chronicity of H. pylori infection [Reference Salama49]. Immune cells that recognize and attack H. pylori accumulate near the site of infection but are ineffective in eliminating the bacterium.
In conclusion, H. pylori infection with multiple HopQ types, truncated cagPAI with increased expression of cagT, LEC and vacAs1 alleles are associated with B-cell NHL in our patients. IHC will be useful in these cases to look for the genetic aberrations associated with H. pylori-independent B-cell NHL.
ACKNOWLEDGEMENTS
Supported by Higher Education Commission of Pakistan grant (No.20-2290/NRPU/R&D/HEC/12) to JY.
AUTHOR CONTRIBUTIONS
Study concept and design – J.Y., Z.A.; acquisition of data – J.Y., Z.u.A., K.M.; analysis and interpretation of data – J.Y., Z.A., K.T., S.A., Z.u.A.; drafting of the manuscript – J.Y., Z.A., S.A., R.K.; critical revision of the manuscript for important intellectual content – J.Y., Z.A., S.A., Z.u.A., R.K., K.M.; statistical analysis – J.Y., S.A., Z.A.; obtained funding – J.Y.; administrative, technical or material support – K.T., J.Y., K.M.; study supervision – R.K., J.Y., K.T.
DECLARATION OF INTEREST
None.




