



Introduction
With 9.9 million new cases in 2019, and 500 000 multi-drug resistant cases, tuberculosis (TB) is far from being eradicated [1]. Among 9 acknowledged lineages (L1 to L9) described in Mycobacterium tuberculosis complex, the lineage L2 is of great interest [Reference Devaux2–Reference Merker6]. Very large outbreaks in L2 were shown to have independently emerged worldwide [Reference Mokrousov7]. Although L2 origin is suspected to be in China and L2 is predominant in east Asia, its exact place and time of emergence are still highly debated [Reference Luo8–Reference Liu10]. L2 clinical isolates have developed specific virulence and drug-resistance features that contribute to their epidemic success [Reference Merker6, Reference Rad11–Reference Klotoe13]. From an evolutionary standpoint, L2 has developed a high IS6110 copy number lifestyle, that could have fostered a hypermutator phenotype that may have increased the virulence or fitness of some of these isolates [Reference Ford14, Reference Gygli15]. The epidemic burst of L2 was first detected during the 90s and was promoted by historical and geopolitical events such as (1) the fall of the former USSR and changes in China; (2) a poorly individualised TB treatment and medical follow-up of prisoners in these countries; (3) the increase in the global trade share of China [Reference Droznin, Johnson and Johnson16–Reference Nicita and Razo18]. L2 fostered many research studies to understand multidrug resistance emergence and to improve its genetic characterisation [Reference Casali4, Reference Bifani19, Reference Mokrousov20]. This was achieved progressively and through the combination of polymorphic markers analysis such as IS6110-RFLPFootnote 1, MIRU-VNTRFootnote 2, Regions of Difference (RD), hypervariable VNTR loci, and lastly whole-genome sequencing (WGS) [Reference Tsolaki21–Reference Shitikov27].
The L2 lineage was split into two main sublineages, L2.1 (Proto-Beijing) and L2.2 (Beijing), [Reference Shitikov28–Reference Thawornwattana31]. L2.1 was described mainly in south-China, particularly in the Guangxi province and could be as ancient as 30 000 years old, having co-evolved with east Asian populations [Reference Luo8]. Among its characteristics, it harbours the RD105 deletion but not RD207 [Reference Srilohasin32]. Rare L2.1 isolates were shown to have become ultra-drug resistant [Reference Srilohasin32]. L2.2, is defined by SIT1Footnote 3 or variants, and is composed of several sublineages, ancestral and modern ones. L2.2.2 defines the asia ancestral 1 sublineage; L2.2.1 gathers all others [Reference Coll33]. RD181 is specifically deleted in all L2.2.1 sublineages [Reference Shitikov28]. The switch from ancestral to modern Beijing is associated to the presence of at least one IS6110 copy in the so-called NTF region and the presence of mutations in replication-repair-recombination (3R) genes, among which the mutT2 G->C mutation in position 1286766 and the ogt C->T mutation at codon 12 position 1477596, relatively to the MTBC H37Rv reference sequence (NC_000962.3) [Reference Rad11, Reference Shitikov28, Reference Yin34]. Until recently ancestral Beijing included 3 asia ancestral lineages (asia ancestral 1, 2 and 3, AAnc1, AAnc2, AAnc3) [Reference Shitikov28], until a new asia ancestral 4 (AAnc4), was discovered in northern Thailand [Reference Ajawatanawong35]. Modern Beijing strains are responsible of most but not of all of the recent MDR-TB outbreaks [Reference Casali4, Reference Gygli15, Reference Karmakar36].
The large databases built with WGS data allow to develop a precise and comprehensive knowledge of L2 diversity [Reference Casali4, Reference Merker26, Reference Thawornwattana31, Reference Casali37]. Computational genomics now allows to study in-depth both the global and the local L2 history [Reference Merker6, Reference Wan24, Reference Merker26, Reference Srilohasin32, Reference Ajawatanawong35, Reference Kang38–Reference Mizukoshi42]. The evolutionary history of L2 isolates is still debated as is their precise dating of emergence and their geographical origin [Reference Merker26, Reference Yin34]. Luo et al. suggested that L2 could be as ancient as 30 000 years old [Reference Luo8]. Merker et al. estimated the time to the most recent common ancestor (TMRCA) of ~6100 and 5200 years before present (BP) for MIRU-VNTR defined clonal complexes BL7 and CC6 (the most ancient) and ~1500 years BP for CC5 (the most recent) [Reference Merker26]. Liu et al. estimated the coalescence between L2.1/Proto-Beijing and L2.2.2/ancestral Beijing at 2200 BP, and at 1300 years BP for the split between all ancestral Beijing lineages [Reference Liu10]. L2.1/Proto-Beijing expansion would have taken place 900 BP whereas modern Beijing would have appeared only around 500 years ago [Reference Liu10]. Hirsch et al. suggested that east Asian and Philippines human populations carrying distinct MTBC lineages may have split only 240–1000 years ago [Reference Hirsh43].
The geographic origin of the emergence of L2 is as blurry as its dating. According to some authors, north-central and north-east China could have been the initial spreading centre [Reference Mokrousov44]. According to others, based on differences in the prevalence of the ancestral L2 lineages in China and on the higher genetic diversity observed in the south-west province of Guizhou, south-China could be the craddle of L2 [Reference Luo8, Reference Yin34]. Indeed, Guizhou counts 17 ethnical minorities and most of the acknowledged ethnic groups of China are located in this province. More generally, south-east Asia shows a higher human genetic diversity than the north-east Asia [Reference Abdulla45]. Arguing in favour of a south-China origin, the recent description of an ‘asia ancestral 4’ L2 sublineage, was done in the north of Thailand in Chiangrai, inhabitated since the 7th century and peopled by ethnic minorities originating from South-China [Reference Ajawatanawong35].
Skeletal evidence of tuberculosis during the Bronze age was found in Korea and Japan [Reference Suzuki, Fujita and Choi46, Reference Suzuki and Inoue47]. In Japan, one of the main characteristics of the tuberculosis history, especially in aged people that were not vaccinated by BCG, is the presence of still poorly characterised ancestral L2 strains [Reference Iwamoto22, Reference Merker26, Reference Wada39, Reference Mizukoshi42, Reference Wada and Iwamoto48]. MIRU-VNTR diversity had been shown earlier to be quite important in L2 isolates from Japan [Reference Mokrousov44]. Other evidence based on MIRU-VNTR had suggested that some specific ancestral L2 strains could be endemic in Japan [Reference Yokoyama49–Reference Yokoyama52]. Since the publication of these studies, WGS of a few reference isolates were published in Kobe and Osaka however they were not mentioned in the simplified L2 phylogeny [Reference Shitikov28, Reference Wada39].
We are currently developing the TB-Annotator project, a new computational genomics pipeline whose aim is to perform data-mining of MTBC genomic diversity using Sequence Read Archives (SRAs). We studied a set of L2 isolates from central Japan Honshu prefecture, Tochigi [Reference Mizukoshi42]. The 169 clinical isolates of M. tuberculosis we studied were from TB patients diagnosed in 2007 and in 2013 [Reference Mizukoshi42]. WGS data on these isolates was released after analysis using a bioinformatical pipeline, described as ‘CAST’ by Iwai et al. [Reference Iwai53]. Since a comparative genomic analysis of these isolates with other ancestral L2 had not been performed, we included these genomes in our database. We also included SRAs labelled as asia ancestral 4 [Reference Ajawatanawong35]. Using our pipeline, we described the genetic characterisation of a new L2 sublineage from Japan, named asia ancestral 5 (AAnc5), that appears to be exclusive from Japan for the time-being. We also assessed the characteristics of the AAnc4 sublineage described in Thailand and provide an updated global evolutionary framework of the L2 lineage [Reference Shitikov28].
Material and methods
Brief description of the TB-annotator pipeline
The full version of TB-Annotator is going to be released in another article (Senelle et al. in preparation). In brief, regarding the set-up of the platform, the processes are the following: SRAs of interest are selected and kept only provided a certain number of conditions that together reinforce the reliability of the data: they must have read length>75 bp, clean reads file must be at least 100 Mo, and CRISPR could be reconstructed using CRISPRbuilder-TB [Reference Guyeux54]. For each SRA, in addition to reconstructing the CRISPR-Cas region using CRISPRbuilder-TB and apart from collecting NCBI information on the genomes, two scripts successively perform the following tasks: (1) search for SNPs according to reference catalogues (Supplementary Table S1) totalling more than 50 000 SNPs, including drug-resistance related SNPs, phylogenetic SNPs as per 26 studies contributing to SNP-based classification [Reference Merker26, Reference Shitikov28–Reference Freschi30, Reference Coll33, Reference Sreevatsan55–Reference Koster75]; (2) search for additional SNPs in each isolate based on H37Rv reference sequence; (3) look for the presence/absence of H37Rv genes as annotated in mycobrowser (https://mycobrowser.epfl.ch/), (4) look for the presence/absence of deletion regions; (5) identify insertion sites of all known insertion sequences in MTBC. The CRISPR locus is rebuilt semi-automatically using a dedicated and previously published script (43/68/360 Spacers format) with an assignment of a Spoligo-International-Type (SIT) tag; the application produces an ordered list of spacers/repeat with variants and IS6110 insertion sequences if present [Reference Guyeux54, Reference Couvin76, Reference Refrégier, Sola and Guyeux77].
Selected genomes
We downloaded a set of 680 SRAs; these samples were selected to represent TB genomic diversity (L1 to L9) described so far, including recent landmark papers [Reference Napier29, Reference Freschi30]. The list of these SRAs is shown in Supplementary Table S2 (list of 680 SRA including 190 L2 SRA); the database was built to represent all L2 sublineages except for the Pacific RD150 sublineage. From [Reference Ajawatanawong35], we selected 28 SRAs labelled AAnc4. From [Reference Mizukoshi42], we initially included 158 SRAs, however 57 SRAs for which the coverage was either too weak or for which it was impossible to rebuild the spoligotype using CRISPR-builder-TB were discarded [Reference Guyeux54, Reference Refrégier, Sola and Guyeux77].
Bioinformatics and phylogenetical methods
Final scripts allow to produce a phylogeny based on the list of studied characters and using RAxML and SplitsTree [Reference Stamatakis78, Reference Kloepper and Huson79]. All computations were performed on the ‘Mésocentre de Franche-Comté’ supercomputer facilities (141 nodes, 2292 cores, 9,27 To memory, 74,2 CPU Power TFlops, 66,4 GPU TFlops), using adequate commands. Apart from the results shown in the Supplementary Table S3, a final phylogenetic tree is displayed graphically and proprietary python scripts allow interactive queries to be made and results to be shown [Reference Kloepper and Huson79]. The current TB-Annotator version includes 6009 genomes and confirmed our results (Senelle et al. in preparation).
SNP-based classification of L2 sublineages
In order to assign the 680 selected SRAs into known lineages and sublineages, we used the reference list of markers defined in Supplementary Table S1. A simplified Venn diagram shows the classification of the 190 L2 SRAs into 125 ancestral and 65 modern isolates (Fig. 1). SNP-based classification results on the 190 SRA of the L2 isolates, as produced by TB-Annotator are found in the Supplementary Table S3.
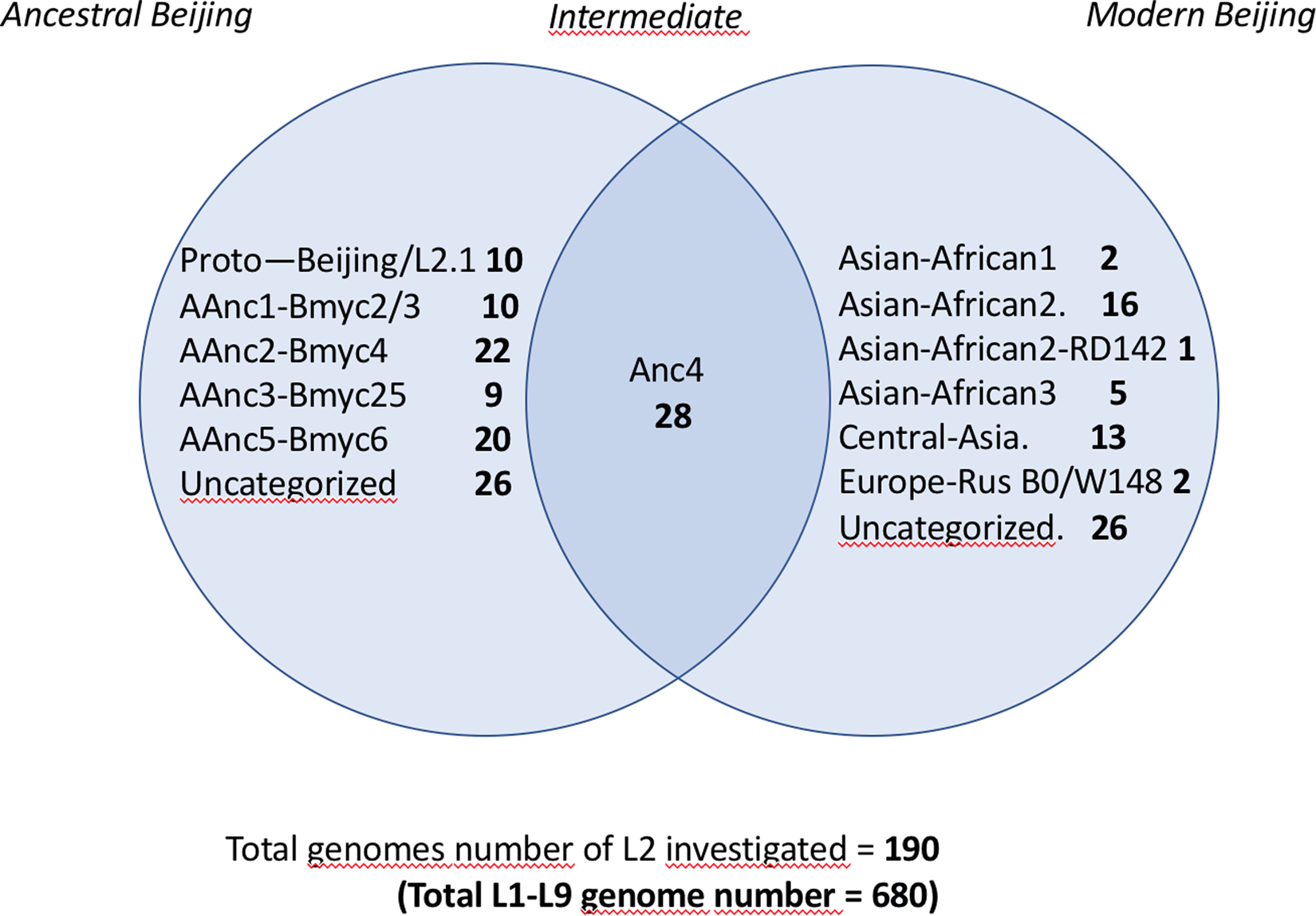
Fig. 1. Venn diagram showing the classification of the 190 L2 SRA studied; note the linking or ‘intermediate’ status of AAnc4 between ancestral and modern strains.
Methods and dataset used to define a new Asia ancestral 5 sublineage (AAnc5)
After the assessment of existing known MTBC sublineages (Fig. 2a and b), the unknown branch (Fig. 2c) was investigated in detail: in Silico CRISPR locus reconstruction using CRISPR-builder TB [Reference Guyeux54, Reference Refrégier, Sola and Guyeux77], in Silico MIRU-VNTR using CAST [Reference Iwai53]; SNPs assessment and intra-cluster distance with TB-Annotator; AAnc5 was shown to originate only from SRAs from Japan isolates (Bioproject PRJDB3875). The bioinformatical pipeline, based on its graphical user interface, allows to select and display new exclusive or shared SNPs and specific genetics characteristics, that were further investigated. Previous genomic markers extracted from Wada et al. 2012 were compared to our results and are found in Supplementary Table S4 [Reference Wada39].

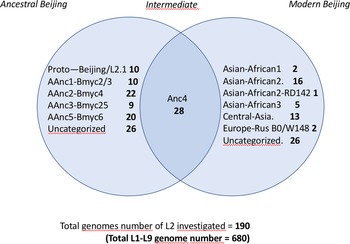
Fig. 2. Left part (a); TB-Annotator unrooted phylogenetic tree on 680 SRA-derived data. L2 samples are shown in blue. Right part (b); close-up on the Lineage 2 with all known branches named except in red the new unknown ancestral Japan sublineage we designated as asia ancestral 5. Central part (c); focus on the unknown Japan lineage.
Results
Discovery of a new L2 sublineage
We implemented a representative set of SRAs of L2 including 101 extracted from the 169 samples study from the Tochigi province study, onto the TB-Annotator platform [Reference Mizukoshi42]. The classification of the 190 L2 studied genomes is shown in a simplified Venn diagram (Fig. 1) and the global phylogenetical tree produced is shown on Figure 2a–c. All samples carried the L2 defining SNP (G497491A) and were found in the same phylogenetic branch (Fig. 2a). Based on SNP search, and as shown on Figure 2b and c and in more detail in Supplementary Table S3, among the ancestral Beijing (n = 125) we found Proto-Beijing (L2.1; n = 10), asia ancestral 1 (L2.2.2-AAnc1, n = 10), asia-ancestral 2 (AAnc2, n = 22), asia ancestral 3 (AAnc3, n = 9), asia ancestral 4 (AAnc4, n = 28), and an unknown branch (suggested as asia ancestral 5 or AAnc5, n = 20); there remained 26 unclassified ancestral Beijing. Figure 1 and Supplementary Table S3.
Ninety-three isolates (including AAnc4), were part of L2.2.1 and were all harbouring a mutT2 G1286766C that is traditionally defining a ‘modern Beijing’ characteristic SNP. Out of these 93, however 65 samples only (excluding AAnc4) were harbouring the second modern L2 signature, i.e. the C1477596 T SNP in ogt, while the 28 AAnc4 did not harbour this SNP [Reference Shitikov28, Reference Ajawatanawong35]. Hence, the designation of modern L2 should be kept for isolates harbouring these two SNPs simultaneously and not only the mutT2 G1286766C polymorphism.
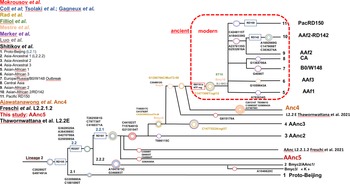
The other modern Beijing (n = 65), are further split into asian african 1 (n = 2), asian african 3 (n = 5), asian african 2 (AAfr2, n = 16), asian african 2-RD142 (AAfr2-RD142, n = 1), central asian (n = 13), Europe/Russia B0/W148 outbreak (n = 2) and there were a remaining 26 unclassified modern Beijing isolates, that did not fit to any described modern sublineage definition, and that were not investigated further in this study. No Pacific RD150 isolates was included in this study. We present an improved unified L2 classification scheme that includes the recent discovery of AAnc4, our own AAnc5, as well as the ‘L2.2.1.2’ [Reference Freschi30], the ‘K’ strain (a member of AAnc1) [Reference Han80], and the L2.2.E [Reference Thomann81], as shown on Figure 3.

Fig. 3. Unified Mtb lineage 2 dendrogram representing the current L2 sublineages with some of their defining SNPs or genetic markers. The colour code tries to superimpose with each author, the size of the circles is arbitrary; this tree tries to provide a simplified unified evolutionary scheme however does not claim to be representative of the full diversity of L2 (redrawn and improved from Shitikov et al., 2017).
The reanalysis of the phylogenetical SNP set described by Shitikov et al. confirmed all the phylogenetical SNPs specific of the ancestral L2 sublineages up to the definition of the modern L2. The mutT2 SNP (G1286766C) is a good phylogenetic marker as it is present only in L2 modern sublineages whereas T1892017C, C4137829T and C4393590G (mutT4) are present in some ancestral sublineages. It is clear from the SNPs results and from the phylogenetical tree shown in Figure 2 that the AAnc4 branch could be called intermediate, as suggested by the team who discovered it, and is neither a bona fide modern L2 sublineage nor a truly ancestral one [Reference Ajawatanawong35]. Conversely, the 20 isolates that we studied from Japan, further designated as AAnc5, all Fulfill the ancestral L2 criteria since they do not possess the expected mutT2 (58) SNP nor any other characteristic of modern Beijing. They also branch before AAnc4. In all cases, the clear-cut split of L2 isolates that either possess none, one, or the two mutT2 (58) and ogt (12) SNPs confirms that these two markers are excellent phylogenetical ones. Based on the results obtained, we thus adapted the unified classification scheme of Shitikov et al. to include some of the new recently described sublineages (Fig. 3). Among those, are the ‘K’ strain (part of AAnc1), the ‘L2.2.1.2’, the L2.2.E [Reference Freschi30, Reference Thawornwattana31, Reference Han80], and the AAnc5 recently designated as L2.2.A [Reference Thawornwattana31].
Description of a new Asia-ancestral 5 (AAnc5) Japan sublineage
Snp-based Results
We further characterized the Japan asia ancestral 5 sublineage (AAnc5) (n = 20) that branches between AAnc1 and AAnc3 (Fig. 2b and c). These isolates can all be defined by many exclusively shared SNPs. The SNP-based classification produced by the TB-Annotator pipeline on the 190 L2 genomes can be found in Supplementary Table S3. In the future versions of TB-Annotator, reports will be downloadable from a dedicated website (Senelle et al. in preparation). The list of 42 exclusive SNPs, found in 16 of these 20 isolates is shown in Supplementary Table S4. An interesting observation is that all the AAnc5 strains harbour a non-synonymous mutation at position 765140 (G->C) in rpoC. Three other genomes (2 in other L2 sublineages and one in a L4 sublineage) also carry this rpoC mutation suggesting independent acquisition.
A pairwise distance matrix between AAnc5 isolates was also computed (Supplementary Table S5); the current pipeline systematically computes the intra-branch SNP distance for clusters or selections of interest below 100 SRAs (results not shown). The pairwise distance between the AAnc5 samples shows a minimum of 166 SNPs (between DRR157280 and DRR157281) and a maximum of 439 SNPs (between DRR130203 and DRR034366) inside AAnc5 isolates, thus excluding recent transmission (Supplementary Table S5). Assuming a mean 0.3 SNP mutation rate per year per genome, these strains might have diverged approximately 250 to 600 years ago from their MRCA. If we accept the suggestion of timing of AAnc4 emergence or expansion around the 7th century in Thailand, (start of Chiangrai), then AAnc5 could have been introduced into Japan earlier, in line with archaeological information [Reference Ajawatanawong35, Reference Suzuki, Fujita and Choi46, Reference Suzuki and Inoue47]. A second list of 46 exclusively shared SNPs, shared between the two most distant isolates on a specific subbranch of AAnc5 (DRR034381 and DRR130203, pairwise distance: 417 SNP) is also shown in Supplementary Table S5.
In silico spoligotyping and reconstruction of the CRISPR locus structure of AAnc 5 using TB-annotator, IS6110 copy number and insertion sites
The twenty studied AAnc5 isolates showed 6 different spoligotype patterns as reconstructed by CRISPR-builder, which was an unexpected result for an L2 sublineage (Supplementary Table S6); most of these patterns have been previously described in the SITVITFootnote 4 database (SIT1, SIT190, SIT269, SIT1364, SIT1674), however one remained undefined as SIT”X”. No SNP variants were found in spacers and repeats, but three isolates exhibited duplications: a duplication of sp65 for DRR034478 and SRR130160, and of sp50 for DRR034476 (Supplementary Table S6). The phylogeny that can be derived from the reconstruction of the CRISPR-Cas structure confirmed the SNPs results: it reveals sporadic deletions of cas genes, Rv2807c, Rv2808c and Rv2813c in some isolates (Supplementary Table S7).
AAnc5 strains were harbouring from 14 to 22 IS6110 copies, and two specific copies were found in almost all these L2 isolates and not in other L2 sublineages: one copy was found at position 1724419 in Rv1527c (found in 15 of these isolates) and the second one was found at position 2041756 (found in 19 of these isolates) (Supplementary Table S6). DRR034455, DRR034471 and DRR034476 were showing the same CRISPR structure, however harboured different missing genes (see next paragraph). Using TB-Annotator, 14 of the AAnc5 isolates were predicted to be drug-sensitive and four were harbouring mono-resistant mutations, two were MDRs (Supplementary Table S8).
Missing genes
Six TB isolates among the 20 AAnc5, apart from showing classical deletions (RD105, RD207, RD181 and PhiRv1), were harbouring specific missing genes: as an example, DRR034363 had Rv1081 to 1084c deleted, DRR034416 was missing Rv1523 to Rv1526c. (Supplementary Table S7). These deletions confirm that phylogenetically linked MTB genomes sometimes harbour strain-specific dependent deletions due to recombination events.
In silico VNTR copy number computation using CAST and comparison with other isolates from previous studies
No specific 15 + 9 VNTR signature could be obtained from in Silico VNTR typing using CAST for any of the AAnc5 isolates [Reference Yokoyama52, Reference Allix-Beguec82]. ETRC, QuB26 and QuB4156 could never be in Silico predicted. Depending on SRA quality, between 6 and 20 VNTR could be predicted (Supplementary Table S9). The VNTR results showed slight variation between isolates; eleven VNTR Loci were invariant in this collection (MIRU04, MIRU10, MIRU16, MIRU20, Mtub29, Mtub30, ETRB, MIRU24, MIRU27, Mtub34, MIRU39) whereas nine loci showed variation (MIRU02, MIRU40, Mtub21, QuB11b, ETRA, MIRU23, MIRU26, MIRU31, Mtub39). When comparing with an in-depth VNTR study performed earlier, AAnc5 was shown to belong to M10 or M37 respectively found in Russia and Singapore [Reference Mokrousov44]. When comparing with a set of 5 reference Japanese isolates (A05N056, ID381, 4558, 4994, 4991/M) that were described to represent the main L2 sublineages found in Japan, ID381 was sharing the same VNTR copy number with AAnc5 on 9 loci (Supplementary Table S9) [Reference Iwamoto22, Reference Wada39]. When comparing in Silico VNTR results with previous VNTR results from published studies in Korea, on the ‘K’ strain, known to belong to AAnc1, we retrieved relatively poor similarity [Reference Kang38, Reference Han80] (Supplementary Table S9).
Comparison between TB-annotator and CAST server results on prospective drug susceptibility testing and on spoligotyping results
When comparing the drug-susceptibility testing results obtained using either the TB-Annotator pipeline or CAST, they were identical (Supplementary Tables S8 and Table S9). Identical results were also obtained on the classical 43 spacers-format spoligotype reconstruction with a minor and yet unexplained discrepancy on a single spacer of a single isolate, DRR034366, for which CAST predicted SIT250 whereas TB-Annotator predicted SIT290 (Supplementary Table S9).
AAnc5 is identical to the G3 endemic L2 ancestral strains cluster in Japan
We compared the 42 SNPs table found in the AAnc5 sublineage (Supplementary Table S4) with the ones found in sequences of 5 Japanese reference isolates, compared to the K1-K2 epidemic strain [Reference Wada39]. According to the definition made by these authors 5 L2 subgroups (G1/2, G3, G4, G5/6 and M) could be defined in Japan based on 10 phylogenetically selected SNPs. From the ID381 strain, a member of the G3 genetic group, described for the first time in Kobe and Osaka in 2006 [Reference Wada39], looking at G3 specific SNPs set (Supplementary file of [Reference Wada39]), and comparing it with AAnc5 SNPs set, we concluded that the G3 group was new and did not fit with the former Shitikov et al. classification scheme. Indeed, AAnc1, AAnc2 and AAnc3 were found to respectively match with G1/2, G4 and G5/6 in [Reference Wada39], however no equivalent was found for G3.
By comparing SNPs lists, we found that the Tochigi province AAnc5 cluster of strains was sharing 36 out of 42 SNPs with the G3:ID381 reference strain found in Kobe and Osaka (Supplementary Table S4). 6 SNPs only (C587945T, G765140C, G1202113A, AGGGAG1476812A, G3148446C and G3820365A) were not found in the ID381 strain. We concluded that the Tochigi strains were highly likely to be historically related to the Kobe and Osaka G3 group described in 2006 through the reference isolate ID381. Accordingly, we propose to retain the common SNPs described by Wada et al. and this study as characteristic of the AAnc5 to fit with Shitikov et al. nomenclature. We positioned both AAnc5 and AAnc4 in the global schematic L2 tree [Reference Shitikov28, Reference Ajawatanawong35]. By digging more in-depth into the comparative SNPs list between our study and the former Kobe-Osaka study, we found that DRR034489 was the closest isolate to the G3 ID381 reference sharing 40 more SNP exclusive to the G3 group, whereas another cluster of 3 genomes were more distant but were sharing 15 more SNPs with ID381 (results not shown). As mentioned above, two very distant genomes, DRR034381 and DRR130203 (417 SNPs pairwise distance) where also sharing 46 additional SNPs that were not found elsewhere (Supplementary Table S5).
Discussion
We describe in this paper a historical endemic ancestral sublineage of L2 based on samples collected in central Japan, Tochigi Prefecture, former Shimotsuke province, that we named AAnc5. This sublineage is highly related to the Japanese G3 group defined in 2012 [Reference Wada39] and assumed to be named L2.2.A in a recent review [Reference Thawornwattana31]. Our results strengthens the phylogenetical relevance of this sublineage into the global L2 evolutionary history and shows that it was transmitted historically in several Japanese cities. The chronology of the emergence of this sublineage relatively to other L2 sublineages was positioned in Shitikov's L2 diversification scheme. Its position relative to previously described lineages clearly showed that it should be qualified as ancestral according to the current definition of this terminology and diverged from other Beijing sublineages shortly after AAnc1.
Tuberculosis is very ancient in mankind history, however it is still impossible to definitively date its emergence in Asia [Reference Liu10]. Early TB outbreak history in Japan could be related to migrations of people from the 5th century BC to the 3rd century AD [Reference Suzuki, Fujita and Choi46, Reference Suzuki and Inoue47]. Tuberculosis was known to be present in ancient time in Japan under the name of rôga, that was used in Chinese medicine [Reference Thomann81]. A link between this disease and TB, as known in the western medicine, was described in 1857 by a medical doctor, Ôgata Kôan, 3 years before the opening of Japan [Reference Thomann81]. There are many traces in Chinese medical history texts of a disease that can be identified as tuberculosis [Reference Barberis83]. The historian Fan Xingzhun gave some clues in his brief study on the history of this disease in China [Reference Fan84]. His analysis of Chinese sources leads him to point out that many terms associated with symptoms suggestive of tuberculosis, appear in ancient sources. The Classic of Mountains and Seas (Shanhai Jing 山海經, 4th–3th century BC) describes a remedy to cure the ‘luo 瘰’, i.e. the scrofulae. However Fan Xingzhun admits that these symptoms may not be specific. He notes that other terms suggestive of the disease appear in other ancient sources such as the Classic of Poetry (Shijing 詩經 gathering texts spanning from the 11th to the 5th century BC) or in the Mengzi (孟子 4th century BC), that Su You 蘇游, a Tang Dynasty (618–907) author, holds as synonyms. Fan Xingzhun stresses that these terms (zhai 瘵,mo 瘼,chuanshi 傳尸 (literally: corpse that transmits), shoubing 瘦病 (literally: thinness disease), zhuanzhu 轉注,fulian 伏練, guzheng 骨蒸 (literally: bones filled with hot steam or hot bones) are often associated with a description of states of extreme fatigue and emaciation in early dictionaries and medical books. The ‘zhai 瘵’ ideogram, in particular, that has become one of the most popular to describe, between others, tuberculosis, is defined in the earliest dictionaries as a disease whose main characteristic is weakness, and, later, as a disease that leads the patient ‘to toss and turn restlessly’ until ‘dying of weakness’. While Fan Xingzhun agrees that all these terms may not necessarily correspond to tuberculosis, the historian nevertheless finds it very likely that the evidence he found in a 14th century book describes a case of pulmonary tuberculosis: ‘Under the Song (d.960-1127), Shi Dezun, when he was 50 years old got the exhaustion and weight loss disease, he was tossing and turning restlessly until he became very emaciated’ [Reference Fan84]. Further, Fan Xingzhun hypothesise (p.97) that tuberculosis prevalence was low in ancient times. However in the Ancient Book of Tang (Jiu Tangshu, 舊唐書 941 AD) it is reported that: « under the Wude reign (618–626), in Guanzhong (Shaanxi province), many have the ‘hot bone disease’ 骨蒸 », a testimony that argues in favour of a tuberculosis epidemic. As the term ‘hot bone disease’ (骨蒸) is already mentioned in the medical Canon ‘The Yellow Emperor's Canon of Medicine’ (Huangdi Neijing 黃帝内經, 2BC-2AC), Fan Xingzhun wonders about the possibilities that tuberculosis could have already been epidemic at that time [Reference Fan84].
Going in-depth into the complex historical phylodynamics history of all MTBC lineages was made possible using the TB-Annotator pipeline that analyses more than 50 000 characters including repeated sequences and SNPs [Reference Napier29, Reference Freschi30, Reference van Embden85–Reference Menardo88]. With an increasing publicly available number of SRAs, it becomes feasible to disentangle all the threads between an ancient and recent historical event that shaped today's TB pandemia, and to understand its relation to ancient/modern population migrations [Reference Liu10, Reference Pepperell89, Reference Mulholland90].
Among the set of AAnc5 characteristics, we describe a non-synonymous mutation in rpoC. It is well known that rpoC mutations are compensatory mutations mainly found in epidemiologically successful isolates that also contain specific rpoB mutations [Reference Comas12, Reference Gygli15, Reference de Vos91]. The rpoC mutation could be an adaptative trait that explains the epidemiological success of MDR-TB L2 isolates in outbreaks such as the Central Asian or the B0/W148 outbreak in Russia [Reference Merker6, Reference Li92] and the epidemiological success in Georgian prisoners [Reference Gygli15]. In the central Asian L2 clade, rpoC mutations may arise within the entire gene and these SNPs are found in epistasia with rpoB mutations [Reference Merker6, Reference Comas12, Reference de Vos91]. In our study, the rpoC mutation, except for the six most recent last isolates, was found in drug-susceptible isolates; hence it is difficult to consider such a mutation as adaptative or compensatory, but rather as a phylogenetical marker of AAnc5 [Reference Gygli15]. Since this SNP was not described in the G3 group from Kobe and Osaka [Reference Wada39], it could be interesting in the future to try to search if there is a significant statistical difference between drug-resistance emergence in one or the other cluster group of isolates. A recent paper in prisoners in Georgia showed that compensatory mutations and patient incarceration were two independent factors associated with the increased transmission, that create a ‘perfect storm’ for MDR-TB transmission [Reference Gygli15]. The precise physiological or fitness consequences of this non-synonymous rpoC mutation in drug-sensitive isolates was not investigated in this study but it could have also functional consequences [Reference Comas12]. L2 evolution includes the early evolutionary history of AAnc5 and could be linked to yet unknown mutator effects specific of some L2 sublineages [Reference Rad11].
Dating of strains diversification may also give some clues on the social and demographic conditions that fostered past epidemics. Calibration of the molecular clock is difficult according to samples, time-frames and lineages (between 0.04 and 2.2 SNPs per-genome-per-year) [Reference Menardo93]. Dating can be comforted if an adequate correlation exists between historical, genomic, epidemiological and demographic facts [Reference Mulholland90]. In a former similar study, we tested three scenarios to date the MRCA of an L4.2 sublineage occurring in Japan and Turkey, and confronted historical, anthropological, human genetics, paleopathological and genomics results [Reference Refregier68]. Here, according to the consensus molecular clock of TB on the middle term, the time frame of coalescence of AAnc5 sublineage landmarks could be between 280 and 310 years ago before present for the closest isolates, and 760–800 years ago for the farthest ones. We based this estimate on L4 and not on L2 mutation rates and hence, are very cautious.
The Tochigi province is famous for its great copper Ashio mine, whose exploitation started beginning of the 17th century. One of our estimations of the expansion date of the AAnc 5 is compatible with the opening of the mine in Ashio [94]. Ashio mine could have been one location of AAnc5 expansion and diversification. Looking at the historical spreading dynamics of the AAnc5 group in Japan could be done in the future with the help of Japanese investigators by looking at finer gradients of all prevalent L2 ancestral sublineages found in Japan.
Another more recent origin of Japanese AAnc5 could be the importation of ancestral L2 clones by forced workers from Korea or China who were made to work in the mining industry from 1939 till 1945 [Reference Thomann81]. Indeed 300 000 Koreans and 38 935 Chinese workers, mostly men were forced to work for Japan during these years [Reference Thomann81]. However given the high SNP number accumulated inside AAnc5, this hypothesis seems unlikely. One limitation of this study is that we could not investigate the potential distribution of G3/AAnc5 isolates within all Japan, however our formal proof that G3 and AAnc5 are linked and deeply rooted is the first insight into a complex history. Future study could try to dissecate further, using a combination on recent statistics on the total of epidemic cases, in combination to genomic characterisation and geographical distribution, the global and the local history of L2 ancestral lineages in Japan [Reference Ota, Hoshino and Hirao95].
The existence of a relatively high diversity of isolates specific to Tochigi and Kobe and Osaka, reminds of historical transmission restricted to a circumscribed area. Of course, such a picture of an endemic past outbreak is more easily observed in islands settings as was shown recently in New Zealand in Maori people who are hosting a specific ‘CS’ (Colonial S-type) L4.4 Sublineage [Reference Mulholland90]. Islands are excellent settings to distinguish endemic from imported species and tuberculosis history is definitively linked to human migration and to local and global demographic history [Reference Sola96]. According to the most recent review on L2 population structure based on more than 5000 genomes, AAnc5 or L2.2.A ‘is the most basal clade of L2.2.1 and comprises isolates almost entirely from Japan. The deep- branching structure of L2.2.A is suggestive of a previously unrecognized endemic strain’ [Reference Thawornwattana31]. Since Japan is an island, and a country with a very low current tuberculosis prevalence rate (13 per 100 000 in 2017), it is an excellent setting to identify the historical events linked to past tuberculosis outbreaks [Reference Refregier68]. The increasing number of available genomes allows more and more L2 sublineages to be discovered, however their intimate historical and epidemiological relationships remains to be studied in more detail [Reference Freschi30, Reference Thawornwattana31].
Conclusions
Thanks to TB-Annotator, a new bioinformatical pipeline that analyses large amounts of the information contained in SRAs, we mapped an endemic L2 ancestral sublineage from Japan onto the global MTBC phylogeny, and we designated it as asia ancestral 5 (AAnc5), and showed that it was linked to the formerly described G3 group in Kobe and Osaka, and now designated as L2.2.A. This sublineage now appears together with some among the most recent one's in a unified evolutionary scheme. AAnc5 possesses many specific characters allowing it to be distinct from all other ancestral sublineages described so far in L2. This finding opens new ways of research, to look for the history of L2 in south-east Asia.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268822000048.
Financial support
This study did not receive specific funding. The authors declare that they have no conflict of interest.
Data availability statement
All data are publicly available either as SRA Accession numbers on the NCBI or EBI website, or provided herein. The source code of the TB-Annotator pipeline is will be published elsewhere (Senelle et al. in preparation).



 Open access
Open access