


Case One
History
(Ryan Taylor) A 52-year-old woman was referred to the behavioural neurology clinic for memory issues. Staff at an Alzheimer’s Society meeting she had attended with questions about her risk of developing dementia noted rapid forgetting and repeated questions at the meeting. A Montreal Cognitive Assessment (MoCA) completed that day was 24/30.
She was a high-functioning single mother, but her sister reported 3 years of worsening forgetfulness. Recently, her eldest child started helping with groceries and cooking, and her sister took over her finances. She used lists to keep herself organized more than before and occasionally missed turns on routine road trips. Occasional errors in speech were noted such as asking, “Do I need to struggle that for you?”
Review of systems was noncontributory. Past medical history included chronic musculoskeletal pain, caesarean section, and depression for 2 years. Her medications included a low-dose aspirin, naproxen, and duloxetine. She did not smoke, drink, or use drugs. She completed grade 12. Her last job was as a factory line worker and the year prior she was put on sick leave because of chronic pain.
Her mother developed rapidly progressive dementia at age 43, and died 6 years after onset despite placement of a shunt for presumed hydrocephalus. One maternal uncle died young of unknown causes. Her brother, age 40, died from complications of vascular disease, her sister who was in attendance was healthy, and another sister age 54 may have had some early cognitive changes.
On examination, she was cooperative and well-groomed. She asked the examiner twice where he was from, and inquired repeatedly whether she needed to keep her appointment reminder notice. A complete neurological examination was normal. She scored 20/30 on the MoCA, with impairment on cube copy, trails, delayed recall, naming, fluency, and abstraction (Table 1).
Table 1 Clinical neuropsychological assessment from both patients. Patient one presented to clinic two years before her older sister, patient two
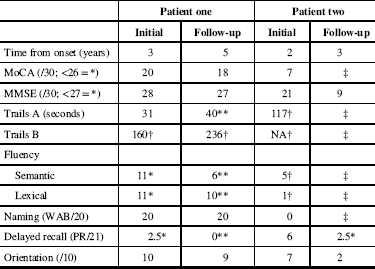
MMSE=Mini Mental State Examination; MoCA=Montreal Cognitive Assessment; PR=prose recall; WAB=Western aphasia battery.
Normative values available: *Consistent with mild cognitive impairment; **Consistent with moderate cognitive impairment; †Consistent with severe cognitive impairment, ‡Unable to complete because of severe cognitive impairment.
Two years later her MoCA score was 14/30 with greater impairment in attention and visuospatial function. Her affect was childlike, and she occasionally embarrassed her daughters by asking strangers personal questions and walking into neighbours’ houses unannounced. She compulsively washed her clothes after wearing them once. She displayed lack of empathy following the death of her father, and attended a Christmas party after witnessing her friend having a fatal heart attack the same night. She had also been admitted to community hospital for an unprovoked seizure, and was started on levetiracetam with good control of her seizures.
Discussion
(Andrew Kertesz) Forgetfulness as a presenting symptom is not very specific, and cognitive symptoms other than memory impairment are often called forgetfulness. Although the early forgetfulness is compatible with Alzheimer’s disease (AD), she has a behavioural disorder that is different from AD. Her affect is childlike and disinhibited. She showed a lack of empathy after the death of her father and her friend, and she adheres to an obsessive-compulsive routine. Furthermore, she has more executive dysfunction than is typically seen in patients with AD who can generally keep up their routine. These are red flags for frontal lobe impairment, possibly frontotemporal dementia (FTD). The frontal lobes regulate our behaviour, inhibition, and social interaction. Alzheimer’s disease is often predominantly parietal, causing difficulty with spatial orientation, but her visuospatial function is preserved; she draws the interlocking pentagons and clock well. My first inclination is to call this FTD; however, there is a family history including her sister and mother, suggesting autosomal dominant inheritance. Her mother also likely had a neurodegenerative dementia, they discovered large ventricles, and she did not improve with a shunt; so, which is more likely to present with and autosomal dominant dementia, AD, or FTD? The game changer is that she developed seizures. Seizures are described only rarely in FTD and neuronal intermediate filament inclusion disease; they are much more common with AD—even more common in presenilin mutations.
Although the clinical pattern is consistent with FTD, my primary diagnosis would be AD with a presenilin mutation. Alzheimer’s disease pathology is seen in ∼20%-30% of clinical FTD presentations. I would test for PS-1, PS-2, and APP. Next on my differential would be mutations for FTD, of which there are many. I would send for routine labs and an MRI. Finally, Tau or amyloid imaging could help differentiate between FTD and AD, respectively, but first want to see if the structural imaging helps.
Follow-up
(RT) Thyroid stimulating hormone, B12, and other routine labs were normal. Serology for HIV and venereal disease research laboratory were negative. Genetic panels for AD (PS1, PS2, APP, APOE4) and FTD (GRN, MAPT, C9ORF72) were negative. MRI completed at her community hospital was reported as moderate diffuse volume loss with enlargement of the frontal horns, and patchy microvascular white matter changes (Figure 1). Three years from initial presentation, she lived in a nursing home because of functional decline, but remained pleasant, ambulatory, and continued to have a normal neurological examination.

Figure 1 MR images from patient one. Sagittal T2 fluid attenuated inversion recovery (FLAIR) images (A–C) demonstrate confluent subcortical and pontine (arrow) white matter hyperintensities and thinning of the subcortical white matter in the frontal lobes. Axial T2 images (D) demonstrate frontal white matter hyperintensities and volume loss, widening of sulci and enlarged ventricles. Midline sagittal T1 weighted images (E) demonstrate callosal thinning that is most pronounced anteriorly. Diffusion weighted imaging demonstrates no areas of restricted diffusion (F), and no parenchymal calcifications are seen on CT (G).
Case Two
History
(RT) Two years after patient one presented to our clinic, her older sister, now age 56, was found to be living in squalor. Garbage piled up in the house attracting insects, her hygiene declined, and she became recluse. At one point she continued to use her toilet even though the water had been shut off for a week and she was noted on several occasions to sit and stare at the TV even though it was off. She demonstrated poor table manners and had significant difficulty using utensils.
She was divorced for many years and lived alone. She completed grade 12 and was working in an accounting firm until the firm closed 4 years ago. Her last job was at a chain restaurant and she quit a year ago. Her past medical history was significant for a lacunar stroke in 1998 causing some mild word finding difficulties. Her vascular risk factors at the time were smoking and oral contraception. She also had depression for several years. Her medications included sertraline, low-dose aspirin, donepezil, levothyroxine, and rosuvastatin.
On examination, she was dishevelled and had flat affect. She easily cooperated with a neurologic examination which was normal. In contrast to patient one’s milder amnestic presentation, she had relative preservation of memory but a total MoCA score of only 7/30 (Table 1). A frontal behavioural inventory performed for both sisters highlights the diametric syndromes of disinhibition and compulsive behaviour in patient one and severe apathy and neglect seen in patient two (Table 2).
Table 2 Frontal Behavioural Inventory

After her initial visit, she began to lose weight despite meals being delivered to her home. She developed rapid cognitive decline with recurrent seizures despite treatment with multiple antiepileptic medications. She died in hospital just 2 years after her first presentation to our clinic.
Neuroimaging
(Sachin Pandey) On axial T2-weighted MRI for patient one (Figure 1), we see some hyper-intense signal in the pons. In the middle cranial fossa, and superiorly there is severe bilateral volume loss in the hemispheres that is most pronounced in the frontal lobes. The distance of the ependymal margin of the lateral ventricles and the overlying cortex is down to a few millimetres. The ventricular calibre is enlarged, which may be germane given her mother’s history of shunt placement. There is less severe volume loss in the parietal and occipital lobes. What little frontal white matter remains is bilaterally demonstrating confluent abnormal signal indicative of underlying white matter disease. There is preservation of ganglionic volume and signal intensity. In the mid-sagittal plane, we see that the corpus callosum is markedly thinned anteriorly worse than posteriorly. There were no focal areas of cystic encephalomalacia or acute/subacute infarcts. There are no masses or foci of abnormal contrast enhancement. In summary, we see bilateral volume loss and confluence of white matter signal abnormality that are most striking in the frontal lobes, with relative preservation of the grey matter volume, and volume of the posterior fossa and brainstem. My impression is that the volume loss is predominantly because of white matter disease although there may be some degree of cortical atrophy. Furthermore, the quality and location of the white matter disease is not typical of microvascular disease. The degree and pattern of white matter abnormality was comparable between the two siblings, if not worse in patient one despite her milder clinical syndrome (Figure 2).
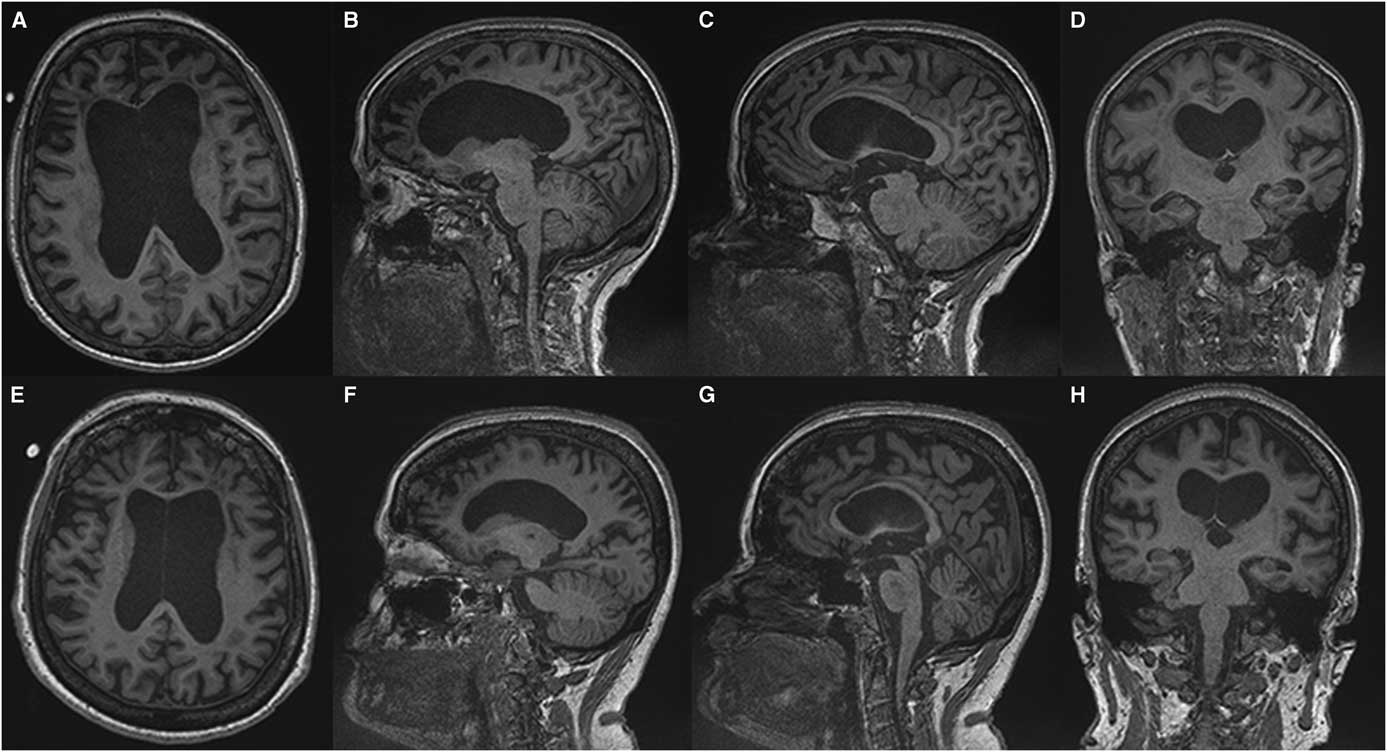
Figure 2 Comparing T1-weighted imaging between siblings. Patient one (A-D) presented with mild to moderate amnestic/executive complaints while patient two (E, H) presented with a rapidly progressive behavioural syndrome. Despite this discrepancy both patients demonstrate a similar degree of atrophy and subcortical volume loss in the frontal and temporal lobes with ventricular enlargement on axial (A, E) and sagittal (C, D) images. A greater burden of frontal white matter hypointense lesions is observed in patient one (A) which belies the relative severity of the her clinical syndrome. Anterior callosal thinning is seen in both patients (E, F).
Discussion
(AK) Although the top diagnosis initially was AD, we now know that there is no AD or FTD mutation. Other genetic mutations to consider in this family would be cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)—the second patient had a stroke, and both have demonstrated striking white matter disease. However, neither patient had a history of migraine and both developed an early clinical syndrome with rapid progression to death in one. This is atypical of CADASIL.
Other conditions associated with white matter changes and huge ventricles include white matter vanishing disease, and the adult-onset leukodystrophies such as hereditary diffuse leukoencephalopathy with spheroids (HDLS) for which causative mutations in the colony stimulating factor-1 receptor (CSF-1R) have been found recently. Another possibility is the hereditary adult-onset neurodegenerative dementia neural ceroid lipofuscinosis. All the above may present with seizures and dementia. I think the most likely diagnosis is going to be HDLS or CADASIL.
Neuropathological Diagnosis
(Basma Alyamany) Autopsy performed on patient two demonstrated atrophy, mainly in the frontal and temporal lobes with dilated ventricles and thinning of the corpus callosum. There is relative sparing of the cortex compared with the degree of white matter loss. Frontal lobe sections stained for myelin show diffuse loss of myelin in the white matter, with sparing of the subcortical U fibres (Figure 3). Neurofilament and amyloid precursor protein staining highlighted axonal swellings (spheroids) in areas of myelin preservation and periodic acid-Schiff staining demonstrated pigmented macrophages. Atherosclerotic disease was seen, and a microcystic infarct was present in the left thalamus. Vessel thickening and perivascular clearing were present in areas of myelin loss. These findings are diagnostic for adult-onset leukodystrophy with adult onset leukoencephalopathy with axonal spheroids and pigmented microglia (ALSP).
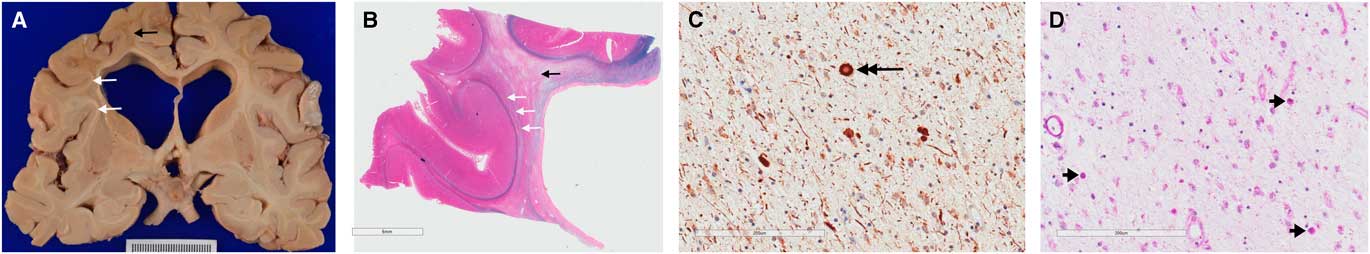
Figure 3 Gross pathological examination demonstrating hallmark findings in ALSP. Gross sections (A) and sections stained with luxol fast blue and haematoxylin and eosin (B) show dilated ventricles with diffuse white matter destruction (black arrows), and subcortical U-fibre sparing (white arrows). High powered views demonstrate axonal swellings, or spheroids, (C) positive for neurofilament stain (large black arrow); and pigmented (D) microglia (short black arrows) on periodic acid-Schiff staining; the later finding is not specific to ALSP.
Genetic Diagnosis
(RT) Genetic testing confirmed the presence of a novel CSF-1R mutation in patient one (NM_005211.3). This finding confirmed a genetic diagnosis of ALSP in this patient.
Concluding Discussion
(AK) This case highlights the importance of adult-onset leukodystrophies in the differential diagnosis of adult-onset hereditary dementias. An interesting feature is the degree of ventriculomegaly in these patients, which would be unusual in AD and FTD. The presence of white matter disease should trigger the possibility of adult-onset leukodystrophies or CADASIL—although I have never seen this degree of atrophy in CADASIL. The fact that CSF-1R mutations affect microglia is considered the likely mechanism by which these patients become demented.
Review
(RT) Adult-onset leukodystrophies manifest variable phenotypes of neuropsychiatric, cognitive, and motoric impairment because of white matter destruction from blood-vessel, axonal or glial dysfunction.Reference Vanderver, Prust and Tonduti 1 Causal genetic mutations continue to surface.Reference Parikh, Bernard and Leventer 2 ALSP has emerged as a unifying diagnosis for pigmented orthochromatic leukodystrophy (POLD) and HDLS. POLD and HDLS demonstrate a spectrum of overlapping histopathologyReference Freeman, Hyman and Sims 3 , Reference Wider, Van Gerpen, Dearmond, Shuster, Dickson and Wszolek 4 and causal mutations in the CSF-1R gene have been identified in both, implicating glial cell dysfunction in the pathogenesis of ALSP.Reference Rademakers, Baker, Nicholson, Rutherford, Soto-Ortolaza and Lash 5 , Reference Nicholson, Baker and Finch 6
ALSP often follows an autosomal dominant pattern of inheritance. However, 40% of CSF-1R mutations appear to be sporadicReference Konno, Yoshida and Mizuno 7 because of de novo mutations and incomplete penetrance.Reference Karle, Biskup and Schüle 8 ALSP cases diagnosed by CSF-1R genotyping are increasing in various populations.Reference Karle, Biskup and Schüle 8 , Reference Wider and Wszolek 9 Common misdiagnoses include FTD, AD, corticobasal syndrome, CADASIL, and multiple sclerosis.Reference Wider and Wszolek 9 Furthermore, CSF-1R mutations likely account for between 10% and 25% of undiagnosed adult-onset leukoencephalopathy cases.Reference Karle, Biskup and Schüle 8 , Reference Guerreiro, Kara and Le Ber 10 Genotype-phenotype correlations are lackingReference Wider and Wszolek 9 and age of onset within families may differ by up to 25 years,Reference Rademakers, Baker, Nicholson, Rutherford, Soto-Ortolaza and Lash 5 suggesting the presence of unidentified modifiers that influence the phenotype and penetrance both within and between families.
ALSP typically presents with early cognitive impairment and neuropsychiatric disturbanceReference Wider and Wszolek 9 because of early, preferential devastation of the prefrontal subcortical white matter.Reference Freeman, Hyman and Sims 3 Although relative preservation of the cortex is seen on histopathology,Reference Freeman, Hyman and Sims 3 , Reference Wider, Van Gerpen, Dearmond, Shuster, Dickson and Wszolek 4 frontal network dementia syndromes follow the disruption of frontotemporal and frontoparietal association fibers.Reference Konno, Yoshida and Mizuno 7 Similarly, disruption of reciprocal cortical connections with deep grey matter structures often causes early Parkinsonism, despite basal ganglia preservation.Reference Sundal, Fujioka and Van Gerpen 11 White matter involvement progresses anterior to posteriorly, eventually disrupting corticospinal and parietal white matter; and motoric impairment manifests as spasticity, gait impairment, ataxia, apraxia, and bulbar dysfunction.Reference Konno, Yoshida and Mizuno 7 Rare cases of cortical blindness in patients with a prolonged clinical course have been described.Reference Freeman, Hyman and Sims 3 Seizures are seen in 30% ALSP cases,Reference Wider and Wszolek 9 despite reports of cortical sparing.Reference Freeman, Hyman and Sims 3 , Reference Wider, Van Gerpen, Dearmond, Shuster, Dickson and Wszolek 4
Comportment frequently mimics the behavioural variant of FTD (bvFTD). In one case series, 27% of patients met the 2011 diagnostic criteria for possible bvFTD, and 11.5% of patients met criteria for probable bvFTD.Reference Wong, Chow and Hazrati 12 Features that distinguish ALSP from bvFTD include the presence of white matter abnormalities on T2-weighted MRI, seizures and EEG abnormalities, a younger age of onset (mean of 42.2 years; range of 15-78), and a rapid progression to severe disability at 3.9 years and death within 6-9 years.Reference Konno, Yoshida and Mizuno 7 , Reference Wong, Chow and Hazrati 12 Amnestic cognitive impairment is also seen in up to 59% of ALSP patients, and 14% of patients present with early memory impairment and behavioural disturbance that is typical of early-onset AD.Reference Wong, Chow and Hazrati 12 Aphasia occurs in 42% of patients,Reference Konno, Yoshida and Mizuno 7 and the non-fluent variant primary progressive aphasia (PPA)Reference Lee, Yun, Jeong, Yoshida, Nagasaki and Ahn 13 and the logopenic variant of PPAReference Oboudiyat, Bigio and Bonakdarpour 14 have been described each in single cases of ALSP.
Bilateral confluent frontal white matter T2-signal hyperintensities with asymmetrical atrophy, enlarged ventricles, and callosal thinning are suggestive of ALSP, but are non-specific findings. Recently described neuroradiologic signs such as cerebral calcifications in an anterior to posterior pericallosal arrangement, seen in 54% of ALSP casesReference Konno, Yoshida and Mizuno 7 , may improve recognition of this syndrome. This pattern of calcification has been reported to predate the clinical syndrome by 10 years,Reference Okamoto, Takeshita, Takahashi, Tanaka, Yoshida and Kuriyama 15 and was seen at birth in a patient who developed ALSP at age 24.Reference Konno, Broderick and Mezaki 16 Furthermore, deep punctate foci of restricted diffusion that enlarge and coalesce over months on MRI are reported exclusively in cases of ALSP, and leukoencephalopathy associated with mutations in mitochondrial alanyl t-RNA synthetase 2 gene (AARS2-L).Reference Lakshmanan, Adams and Lynch 17 AARS2-L is often clinicopathologically indistinguishable from ALSP, but may be differentiated by the absence of cerebral calcifications, autosomal recessive inheritance, abnormal muscle biopsy, and ovarian failure in all women.Reference Lynch, Zhang and Lakshmanan 18
ALSP presents a diagnostic challenge for neurologists across subspecialties. Fortunately, emerging genetic and neuroimaging characterization, and recently published diagnostic criteria,Reference Konno, Yoshida and Mizuta 19 may improve antemortem recognition and diagnosis of this condition. Further research is needed to clarify unknown developmental, environmental, or genetic modifiers that underlie the phenotypic variability demonstrated in this and other reports. ALSP associated with CSF-1R mutations is being recognized increasingly as a cause of hereditary and sporadic early-onset frontal network dementia, and should be considered particularly in the presence of rapid decline, seizures, white matter abnormalities, or other characteristic neuroimaging findings.
Acknowledgements
The authors would like to thank Dr. Robert Hammond for providing guidance and Lisa Baker Spiller for organizing space and time for this Schulich Clinical Neurological Sciences Clinicopathological Conference.
Disclosures
RT, BA, SP, AK, LA, and EF have nothing to disclose.
Statement of Authorship
RT performed the chart review, writing of the manuscript, generation of the figures, and lead the case presentations. BA gave the pathological diagnosis, selected of key images for pathology figures, and discussion of neuropathology. SP reviewed the neuroimaging data and provided discussion of the images. AK performed expert case discussion. LA provided expert review of the cases and pathological discussion. EF performed editing of the manuscript, project oversight, and patient care.






 Open access
Open access