



Treatment-resistant depression
Major depressive disorder (MDD) is a leading source of disability worldwide.1 At least a third of people with MDD fail to respond to an adequate trial with first-line antidepressants and hence have varying degrees of treatment-resistant depression (TRD).Reference Rush, Trivedi, Wisniewski, Nierenberg, Stewart and Warden2, Reference Carvalho, Berk, Hyphantis and McIntyre3 Treatment resistance significantly contributes to the global burden of depression.Reference Rosenblat, McIntyre, Alves, Fountoulakis and Carvalho4 Relatively few pharmacological treatments are currently available for TRD.Reference Rosenblat, McIntyre, Alves, Fountoulakis and Carvalho4 Therefore, the investigation of mechanistically novel neurotherapeutic targets (i.e. beyond standard monoamine-based approaches) for TRD is a clear unmet need in the field.Reference Rosenblat, McIntyre, Alves, Fountoulakis and Carvalho4
Statins
Statins are pleiotropic agents and accumulating evidence from preclinical studies has suggested that these agents may have antidepressant properties.Reference Rosenblat, McIntyre, Alves, Fountoulakis and Carvalho4, Reference Ludka, Constantino, Kuminek, Binder, Zomkowski and Cunha5 A recent population-based study indicated that concomitant use of statins and selective serotonin reuptake inhibitors (SSRIs) resulted in significantly less psychiatric hospital contacts because of depression compared with individuals who used SSRIs alone.Reference Köhler, Gasse, Petersen, Ingstrup, Nierenberg and Mors6 Moreover, a meta-analysis of population-based studies has suggested that statin use may decrease the risk of incident depression.Reference Parsaik, Singh, Murad, Singh, Mascarenhas and Williams7 In addition, a recent meta-analysis of three small randomised controlled studies (RCTs) have provided evidence that adjunctive treatments with statins could be efficacious for treatment of depressive symptoms in patients with moderate-to-severe depression.Reference Salagre, Fernandes, Dodd, Brownstein and Berk8
Several mechanisms may contribute to the putative antidepressant mechanisms of statins including anti-inflammatory, antioxidant, neurotrophic and even monoamine-based effects.Reference Ludka, Constantino, Kuminek, Binder, Zomkowski and Cunha5, Reference Köhler-Forsberg, Gasse, Berk and Østergaard9–Reference Herbet, Izdebska, Piątkowska-Chmiel, Poleszak and Jagiełło-Wójtowicz11 Aberrations in these pathways have been increasingly implicated in the neurobiology of depression.Reference Moylan, Berk, Dean, Samuni, Williams and O'Neil12–Reference Köhler, Freitas, Stubbs, Maes, Solmi and Veronese14 Moreover, abnormalities in lipid metabolism have also been implicated in the pathophysiology of depression, specifically for the development of atypical depressive symptoms i.e. increased appetite and/or weight gain, hypersomnia, leaden paralysis, fatigue and interpersonal sensitivity.Reference Parekh, Smeeth, Milner and Thuret15, Reference Lamers, Milaneschi, de Jonge, Giltay and Penninx16 Hence, the lipid-lowering effects of statins may also play a role in the putative antidepressant effects of this class of drugs. Data from the STAR*D trial indicate poorer treatment response to SSRIs in those with atypical depression and therefore, statins could provide genuinely novel therapeutic targets for this group of people with TRD.Reference Stewart, McGrath, Fava, Wisniewski, Zisook and Cook17
Aims of the RCT
We plan to carry out an RCT involving 150 participants with TRD, with the aim of determining whether the addition of simvastatin (20 mg daily), a lipophilic statin that readily crosses the blood–brain barrier.Reference Johnson-Anuna, Eckert, Keller, Igbavboa, Franke and Fechner18, Reference Thelen, Rentsch, Gutteck, Heverin, Olin and Andersson19 to treatment as usual (TAU) for 12 weeks leads to a reduction in depressive symptoms compared with placebo added to TAU. We predict that patients with TRD will show improvement in mood with simvastatin treatment and that the response will be associated with reduction in lipid biomarkers (i.e. plasma low density lipoprotein (LDL) and LDL/low density lipoprotein (HDL) ratio).
A secondary hypothesis relates to simvastatin's effects on putative inflammatory biomarkers. High LDL and low HDL contribute substantially to immunological upregulation in the development of atherosclerosis. Proinflammatory cytokine (interleukin-6 and tumour necrosis factor) levels are often altered in patients with depression and are decreased by successful antidepressant therapy.Reference Ma, Zhang and Baloch20, Reference Liu, Ho and Mak21 We therefore predict that in those patients with TRD who show improvement in mood with simvastatin treatment that response will be associated with reduction in inflammatory biomarkers. Our mechanistic hypothesis is that the anti-inflammatory and lipid-reducing effects of simvastatin will improve neuroinflammation and thus mediate a response to simvastatin.
Method
This will be a pragmatic multicentre, 12-week, double-blind, placebo-controlled randomised trial of simvastatin added to TAU for patients with a DSM-5 (as measured by SCID-5)Reference First, Williams, Karg and Spitzer22 major depressive episode that has failed to respond to at least two trials of antidepressants. It will be a parallel group study with 75 participants in each arm. Recruitment will occur at out-patient psychiatric clinics in Karachi, Lahore, Hyderabad, Quetta and Rawalpindi, Pakistan. Consistency of procedures across sites will be ensured through vigorous training and supervision of researcher staff. This will include: ensuring research team members involved in the data collection and recording are centrally and consistently trained in the correct processes; having checklists for tasks involved in data collection; providing all researchers involved in assessments with clear, written instructions through a manual of operations and procedures/standard operating procedures, which will detail operational data definitions, recruitment, screening, enrolment, randomisation, follow-up procedures, data collection methods, data flow, case report forms and quality control procedures.
Participants will be randomised to receive either simvastatin and TAU, or placebo and TAU for 12 weeks. All participants will provide written informed consent after reading the information in English and/or Urdu. TAU will comprise medication including antidepressants, mood stabilisers and antipsychotics and routine input from mental health services. There are little, if any provisions for psychosocial interventions for patients with depressive disorders in Pakistan. Simvastatin added to TAU will be 20 mg taken once daily. This dose has been found to be safe and well tolerated as well as effective in reducing depressive symptoms in a population with moderate-to-severe depression.Reference Gougol, Zareh-Mohammadi, Raheb, Farokhnia, Salimi and Iranpour23
Sample size
We estimate a moderate effect size of 0.60 will be reported at week 12. Assuming a type I error of 5%, we will need to follow-up 120 patients across the two groups to achieve 90% power at week 12. With an anticipated drop-out rate of 20% by week 12, we will randomise 150 participants. An effect size of 0.4 or greater for difference in Montgomery–Åsberg Depression Rating Scale (MADRS)Reference Montgomery and Asberg24 scores in RCTs of antidepressants versus placebo has been suggested as necessary to suggest clinical utility for an antidepressant used as monotherapy.Reference Jakobsen, Katakam, Schou, Hellmuth, Stallknecht and Leth-Møller25 A sample of 120 participants is therefore powered to detect moderate to large effect sizes and trends towards significance in areas of smaller effect.
Study procedures
Recruitment
Initially, the research team will approach local clinical teams to inform them about the trial and provide them with the inclusion and exclusion criteria. They will then ask psychiatrists in each out-patient department if they are able to identify any patients who may be potentially eligible to take part in the study. The consultant psychiatrist will briefly explain the study to the patient and if the patient agrees, consultants will refer patients to the research team (Fig. 1).
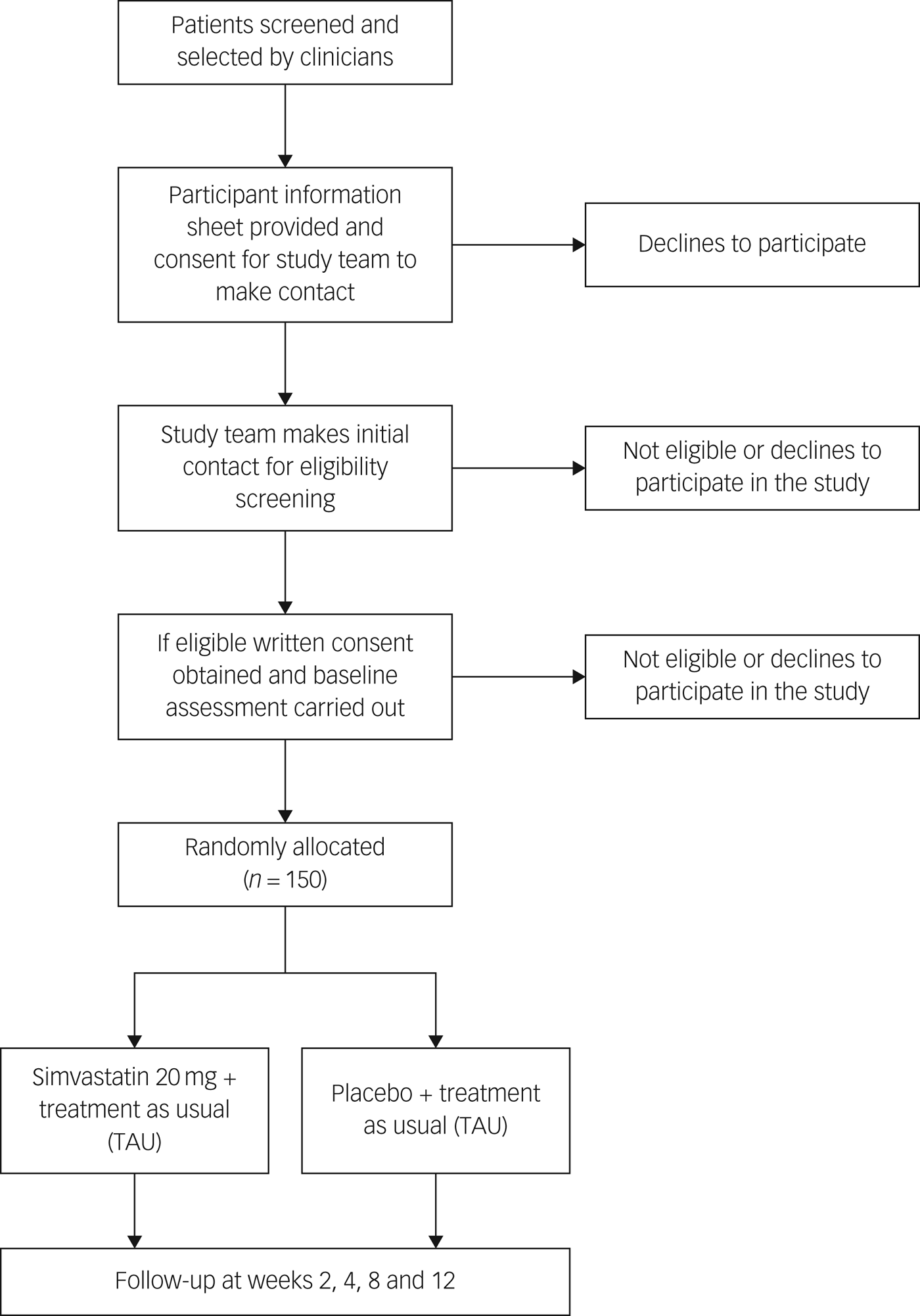
Fig. 1 Flow chart of randomised controlled trial.
Researcher staff will work closely with the clinical team to determine if patients are suitable for participation in the trial. If patients meet the inclusion criteria, and the consultant psychiatrist and clinical team agree that they could be potential participants, the research assistant will arrange an appointment to explain the study verbally and provide them with the participant information sheet. The patient will be given at least 24 h to read and understand this leaflet. If after this they decide that they are willing to take part, an appointment will be arranged to obtain signed informed consent for trial participation and also signed consent for access to their medical notes. Literate participants will sign the consent forms but, if the participant cannot read and sign the consent form, his/her caregiver and/or an independent person who the participant agrees to be a witness, will be requested to read the participant information leaflet and consent form to the patient. Participants will then be asked to place a thumbprint on the consent form if they agree to participate, which will be countersigned by a witness/caregiver.
Screening
At the first visit, participants will undergo structured diagnostic interviews using the Structured Clinical Interview for DSM-5 (SCID-5) to confirm a diagnosis of DSM-5 current major depressive episode.Reference First, Williams, Karg and Spitzer22 This tool has been validated for use in the Urdu language and has been used in previous studies in Pakistan.Reference Husain, Chaudhry, Husain, Khoso, Rahman and Hamirani26 The 24-item Hamilton Rating Scale for Depression (HRSD-24) will be used to assess severity and a score ≥14 will be used as the minimum threshold for study entry.Reference Hamilton27
All patients must currently be on an antidepressant and must have had a non-response to more than two oral antidepressant treatments in the current episode (including the one they are currently taking). The more than two antidepressants must have been taken for at least 6 weeks at least at the minimum therapeutic dose according to British National Formulary (BNF)28 and Maudsley prescribing guidelines.Reference Taylor, Barnes and Young29 Relapse while on an antidepressant will also count as a failed treatment trial. During the 12 weeks of active/placebo treatment patients will be requested to remain on a stable dose of the antidepressant they are taking. Participants will not be permitted to start a psychosocial intervention or psychotherapy during the study period; however, those who were already engaged in these treatments at the screening stage will be permitted to continue their treatment.
Inclusion criteria
The inclusion criteria for participants are men and women aged 18–75 years with a diagnosis of major depressive episode confirmed by SCID-5 and two or more failed trials of antidepressant medication, at minimum effective doses (as per the BNF and Maudsley Prescribing guidelines) for at least 6 weeks, who are in contact with mental health services. Participants must be able to demonstrate the capacity to provide informed consent as assessed by their own clinician and able to complete the required evaluations and take oral medication. Effective contraceptive precautions (either the use of barrier methods or the oral contraceptive pill) are to be taken by women of child-bearing age; a negative pregnancy test will be required in order to meet inclusion criteria.
Exclusion criteria
Exclusion criteria were a primary psychotic disorder or bipolar disorder; history of intolerance to statins or presence of any contraindication to statins; presence of any serious medical condition or neurological problem; presence of an autoimmune or inflammatory disorder (for example systemic lupus erythematosus, rheumatoid arthritis and inflammatory bowel disease); alcohol or drug dependence; active suicidal ideation; pregnant or breastfeeding. Patients with a serum LDL level of <80 mg/dL at baseline will also be excluded from the study as are patients with abnormal hepatic enzymes (aspartate transaminase (AST) and alanine transaminase (ALT)) or abnormal lactate dehydrogenase (LDH) or creatine phosphokinase (CPK) values at baseline. Patients already on statin treatment will also be excluded.
The criteria for leaving the study are: (a) at the participant's request; (b) at the discretion of the responsible physician or trial investigator (for example an adverse event, poor adherence). Poor adherence is defined as either taking <75% of study medication between assessments points at baseline, week 2, 4, 8 and 12 or missing trial medications all together for ≥7 days in the period; and (c) pregnancy.
Randomisation and allocation concealment
The randomisation service will be provided by an independent statistical support service based in the UK. We will stratify randomisation based on severity of depressive symptoms at baseline and the study centre. Participants will be assigned to groups via random generation of allocation sequence, balanced across allocation group. Each participant will be assigned a unique study patient identification number once they have given informed consent and eligibility has been confirmed. The central trial pharmacist will prepare a 12-week package of treatment bearing the patient's name and ID number and send it to the site pharmacy. Thus, the site pharmacy will not know the treatment allocated of the patient. A study information leaflet will be given to the participant, explaining that they are in a clinical trial and that in addition to TAU they are taking a placebo or simvastatin. This leaflet will also have the name of the local principal investigator.
Allocation will be masked from study investigators and coinvestigators until participants have completed all follow-ups and the database is cleaned and locked. The trial pharmacist at the central pharmacy will keep the drug codes. There will be detailed protocols regarding maintenance of masking for raters and governing unmasking procedures in cases of emergency for pharmacists. To assess the integrity of masking procedures, participants and independent raters will be asked to complete a conventional guess form asking whether they believe participants received simvastatin or placebo as a treatment after the final ratings have been completed.
If any participant develops side-effects or has an emergency requiring drug unmasking, the responsible consultant and care team will be informed of the trial drug, probable side-effects and make a decision about participant continuation in the trial. Participant well-being and safety will be crucial at all times and locally and internationally accepted Good Clinical Practice will be observed across the trial's governance procedures.
Follow-up
Participants will continue with the treatment currently provided by their clinical teams. Simvastatin will be started at a dose of 20 mg daily. Assessments will be at baseline, weeks 2, 4, 8 and 12 (see Appendix). The clinical team and consultant psychiatrist will continue to oversee routine care for each participant although research assistants will be contactable for the duration of the study to respond to any concerns. Demographic data will be collected by research assistants at baseline and will include age, gender, marital status, socioeconomic status, highest level of education, smoking status and concomitant medication.
Participants will be asked to bring all leftover medications to each follow-up visit, so that research assistants can complete a pill count to monitor adherence. On each visit the research assistant will meet with the patient at an agreed location to complete the follow-up assessment.
Outcome measures
The primary outcome measure will be the MADRSReference Montgomery and Asberg24 total score at week 12. Ratings will be made on the basis of a semi-structured clinical interview at baseline and at every follow-up visit until week 12. The MADRS was chosen as the primary outcome measure as it has been demonstrated to be more precise in estimating changes in depression severity compared with other measures.Reference Carmody, Rush and Bernstein30, Reference Carmody, Rush, Bernstein, Warden, Brannan and Burnham31 However the HRSD remains a useful screening tool and the use of different assessment scales at screening and follow-up mitigates the risk of clinician bias whereby raters overestimate symptom severity at the screening stage to increase study recruitment. This can then mask the true treatment effect and may lead to an inflated placebo response.
Other outcomes recorded will include: the HRSD-24,Reference Hamilton27 which includes assessment of atypical depressive symptoms; the Clinical Global Impression scale,Reference Busner and Targum32 an overall measure of illness severity; the 7-item Generalized Anxiety Disorder scale,Reference Spitzer, Kroenke and Williams33 a self-report measure of severity for generalised anxiety disorder; and a modified Morisky measurement of medication adherence.Reference Morisky, Green and Levine34 We will also assess changes in body mass index from baseline to week 12. All scales have been validated for use in the Urdu language and have been used in previous studies in Pakistan.Reference Husain, Chaudhry, Husain, Khoso, Rahman and Hamirani26 Adverse effects will be monitored using the UKU rating scale.Reference Lingjaerde, Ahlfors, Bech, Dencker and Elgen35
Measurement of biomarkers
Participants will be asked to provide two blood samples: at baseline and week 12. We aim to analyse the relationship of simvastatin to lipid and inflammatory markers and determine if this is associated with symptom change prior to a clinical effect (if detected). The biomarkers analysed will include lipid biomarkers HDL, LDL and c-reactive protein (CRP), a peripheral marker of inflammation.
Interrater reliability
Research assistants have been trained in the use of SCID and in carrying out clinical assessments by the University of Manchester for a previous grant funded study. Interrater reliability will be measured using calculations of the kappa statisticReference Fleiss36 assessed during investigator meeting rating precision exercises.
Statistical analysis
Initial descriptive analysis will be conducted to study the profile of the participants and investigate group differences at baseline on main demographics and clinical measures. Frequencies and proportions will be used for categorical variables and mean and standard deviation, median and interquartile range, minimum and maximum for continuous variables. No statistical significance tests will be used to compare groups. Drop-out, visit frequency and adherence to treatment will be summarised by group with the appropriate descriptive statistics.
Primary analysis
The main analysis will follow the intention-to-treat principle in which participants are assigned to treatment groups as randomised. Participants that drop-out of the study will be included in the analysis provided that they have a baseline score for the outcome. For the primary hypothesis, the MADRS scores at week 12 will be compared between groups using a mixed-effect model. The outcome for the model will be all post-baseline MADRS scores and the fixed effects will include baseline MADRS scores, treatment group, time (post-baseline time points) and a treatment group × time point interaction. To account for the dependencies between repeated measures on the same patients, an intercept for each individual will be included as a random effect.
A linear contrast will be used to test the primary hypothesis, the difference between groups at week 12 in MADRS scores. Given the modest sample size it will likely not be possible to use as random slope for each participant; however, the covariance matrix of the residuals of the model can be allowed to correlate to account for dependencies across time. Two-tailed tests and an alpha level of 0.05 will be used for significance. All estimates will be presented with 95% confidence intervals. To give an estimate of effect size, standardised regression coefficients will be calculated using the overall standard deviation of the baseline outcome scores as the metric. One advantage of this approach is it gives the magnitude and direction of effect in units of patient population of interest.
Secondary analyses
Secondary analyses will use similar mixed models to that of the primary outcome, baseline adjusted with time, treatment group and the interaction between treatment group and time as independent variables. As for the primary outcome, the main time point of interest is 12-week follow-up and treatment effects will be determined by contrasts at the measured time point. To compare response and remission rates between groups logistic regression will be used in which patients will be classified as in remission or not according to standard criteria on the MADRS scale. As there are multiple measures, P-values will be adjusted for multiple comparisons using a correction based on controlling the family-wise error. Outcomes at time points other than 12-week follow-up will be considered exploratory and the main focus will be effect sizes (standardised regression coefficients).
To compare the frequency of adverse events descriptive summaries will be presented by treatment group. Differences in the rates of adverse events will be assessed by a generalised linear mixed model. Poisson or negative binomial distribution will be used given sufficient numbers of events, where time, treatment group and their interaction are fixed effects with participants as random effects. Alternatively, simple count models will be used at each time point given the limited sample size and complexity of generalised linear mixed models.
Missingness
It is likely there will be some drop-out of participants from the trial. Baseline predictors of missingness can be identified using logistic regression with the outcome (missing or not). Mixed-effect models fitted through maximum likelihood can use all available information in the data potentially avoiding removal of participants who have dropped out from the analysis. Moreover, by including baseline predictors of missingness in the statistical models, the analysis may assume the data to be missing at random. Diagnostic analysis will be conducted through checking residuals for outliers, influential data points and normality. If outliers or influential points are found a sensitivity analysis will be conducted after removing such points to see if the main findings change.
Analysis of exploratory hypothesis
The exploratory hypothesis that baseline lipid/CRP levels will affect response to simvastatin will be tested by adding the baseline lipid/CRP to a model for the primary outcome at 12 weeks, adjusted for baseline and including a treatment group × baseline lipid/CRP interaction. The lipid/CRP level will be considered moderators if the interaction with treatment group is significant (at a significance level of 0.05, using bootstrapping for inference and construction of confidence intervals). If this is the case, exploratory plots that look at the group effect at different levels of the moderators will be used to study the nature of the moderation.
In order to test the hypothesis that changes in peripheral levels of lipids/CRP will be a mediator in the path between treatment and change in MADRS scores, an initial descriptive analysis will be conducted to look at the bivariate association between treatment group and change in lipid/CRP levels, and between change in lipid/CRP (at 4 and 12 weeks) and change in MADRS scores. A mediation model will then be fitted to the data that jointly models the effects of treatment on the change in lipid/CRP at 4 weeks and MADRS at 12 weeks. The mediation effect is tested by the proportion of the total effect from treatment group to change in MADRS that goes through change in lipid/CRP. This model will be fitted in Lavaan in R or Mplus 7.11Reference Muthén and Muthén37 and the indirect effect estimated through bootstrap resampling. Given the sample size, the effect sizes for the indirect mediation pathway would need to be relatively large.Reference Fritz and MacKinnon38
Safety monitoring
Adverse events monitoring will be undertaken from the time informed consent is obtained until follow-up at 12 weeks (using the UKU scale). Laboratory parameters including total serum cholesterol, LDL, HDL, triglyceride, AST, ALT, LDH and CPK will be checked by the study physician before patient entry into the trial, and if there are any safety issues what mean the patient should not be started on the study medication that individual will not be included to the study.
Study coordination
Local investigators will chair a weekly meeting with research assistants to help coordinate the study. The chief investigator will hold meetings with the research team every 2 weeks via Skype.
Data protection and confidentiality
For data security, consent forms and paper copies of assessment tools that have any identifying information on them such as name, address or phone number of participants will all be stored in locked filing cabinets in a secure office. All computerised data will be encrypted and password protected. We will also maintain quarterly site audits of the data and security protocols across each site. These audits will be independent from the investigators and the sponsor.
Trial steering committee
The responsibility of the trial steering committee will be to offer the overall supervision and monitoring of conduct of the trial. The committee will be independent of the investigators, their employing bodies, funders and sponsors. It will monitor overall trial progress and conduct, and will also advise on scientific credibility. The trial steering committee will reflect and act, as suitable, upon the recommendations of the data safety and monitoring board (DSMB) for deciding if the trial needs to be stopped on grounds of safety or efficacy.
DSMB
National Institutes of Health guidelines will be followed for the DSMB for monitoring and conduct of the study. The study team will be accountable to an independent DSMB, which will comprise independent members including a chair with expertise in clinical trials, a biostatistician, one clinician with expertise in efficacy monitoring and one clinician with expertise in safety monitoring. The board will meet once a year and when necessary. The DSMB will be the only body having access to the unmasked data. The DSMB will make its recommendations to the principal investigator and trial steering committee.
Declaration of Helsinki
The clinical trial will be conducted according to the guidelines of the Declaration of Helsinki (1974) as revised in Tokyo (1975), Venice (1983), Hong Kong (1989), South Africa (1996) and Scotland (2000). The research team will comply with the International Conference on Harm minimisation/Good Clinical Practice Guidelines (1996) which are in accordance of the principles of the Declaration of Helsinki.
Timeline
The anticipated start date for recruitment is January 2019 with a total project duration of 36 months.
Results
The trial was registered with ClinicalTrials.gov (identifier: NCT03435744) on 16 February 2018 (https://clinicaltrials.gov/ct2/show/NCT03435744).
Ethical considerations
The National Bioethics Committee of Pakistan has provided institutional review board approval for the study.
Discussion
At least a third of patients with MDD have treatment resistant depressionReference Rush, Trivedi, Wisniewski, Nierenberg, Stewart and Warden2 and up to a third of patients also show evidence of an activated inflammatory response.Reference Wium-Andersen, Orsted and Nielsen39 There is increasing evidence linking immune–metabolic disturbances to MDD. Numerous studies have demonstrated the anti-inflammatory effects of statins in altering cytokine release and phagocytic activity.Reference Benati, Ferro, Savino, Ulivieri, Schiavo and Nuccitelli40–Reference Djaldetti, Salman, Bergman and Bessler45 Simvastatin has been shown to be superior to alternative statins in preventing neurodegenerative conditions, because of its permeability across the blood–brain barrier and ability to prevent cell death.Reference Sierra, Ramos, Molina, Esteo, Vázquez and Burgos46 Moreover, TRD has been shown to be a neuroprogressive conditionReference Setiawan, Attwells, Wilson, Mizrahi, Rusjan and Miler47 yet despite clinical trials demonstrating simvastatin's potential efficacy as an adjuvant treatment for MDD,Reference First, Williams, Karg and Spitzer22 we are unaware of any clinical trials in TRD.
To our knowledge, the current trial will be the first trial investigating the use of simvastatin as an augmentation strategy in patients with TRD. If this study indicates that adjuvant simvastatin is efficacious in reducing depressive symptoms, it will deliver immediate clinical benefit. It is an inexpensive, off-patent drug that is readily available worldwide. It could potentially be a cost-effective treatment option in settings with diminishing resources such as low- and middle-income countries as well as other publicly funded healthcare services. The results of the biomarker analysis may provide insights in to the mechanism of action of simvastatin and if reductions in plasma lipids and/or inflammatory biomarkers are associated with an individual response to adjuvant simvastatin, it could inform the development of a ‘precision medicine’ approach to the treatment of TRD. Furthermore, the results of this study will inform the current evidence base of treatment options for patients with treatment-resistant depressive symptoms, a condition that causes significant disability and functional impairment yet for which there is a clear paucity of controlled clinical trials of novel treatments.
Funding
This study has been funded by the Pakistan Institute of Living and Learning. It represents independent research partly funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR or the Department of Health.
Appendix
Table A1 Assessment schedule




 Open access
Open access
eLetters
No eLetters have been published for this article.