


1. Introduction
The need to restrict the increase in the rate of inbreeding (ΔF) in genetic improvement programmes has been widely recognized. Restricting ΔF allows restriction of the decrease in genetic variability and, most importantly, the reduction in fitness-related traits. Over recent years, selection methodologies (‘optimized selection’) have been developed to maximize genetic gain whilst constraining ΔF by optimizing the contributions made by parents to the next generation (Meuwissen, Reference Meuwissen1997; Grundy et al., Reference Grundy, Villanueva and Woolliams1998). Although these methodologies were developed assuming the infinitesimal genetic model, they are also effective for mixed inheritance models where the identified QTL are known to be segregating with the polygenes (Villanueva et al., Reference Villanueva, Pong-Wong and Woolliams2002).
A key component of optimized selection is the numerator relationship matrix, as the selection method takes into account all genetic relationships between selection candidates together with their estimated breeding values. The use of the numerator relationship matrix conditional on pedigree (Ap) to restrict global coancestry in the optimization tool ensures maintenance of ΔF and genetic variation at the desired level for loci unlinked to those under selection but not for selected loci or loci linked to them (Villanueva et al., Reference Villanueva, Pong-Wong, Fernańdez and Toro2005).
It may be desirable to maintain genetic variation in specific genome regions close to loci under selection. An example would be the MHC region where loci affecting traits selected by breeders (e.g., performance traits) might be located. Bot et al. (Reference Bot, Karlsson, Greef and Witt2004) suggested that loci affecting clean fleece weight are segregating in the MHC region in sheep. In pigs, the SLA region on chromosome 7 has possible associations with many production and reproduction traits (Vaiman et al., Reference Vaiman, Renard, Borgeaux, Warner, Rothschild and Lamont1988). QTL detection studies have reported back fat thickness effects between −3 and +3·7 mm for specific SLA haplotypes (de Koning et al., Reference de Koning, Janss, Tattink, van Oers, de Vries, Groenen, van der Poel, de Groot, Brascamp and van Arendonk1999). A restriction on ΔF at a specific region in the genome could be achieved by applying optimized selection using a numerator relationship matrix constructed using pedigree and molecular markers located in and around that region.
The study aims to assess (i) the effect of optimized selection (using a numerator relationship matrix based on pedigree only) on genetic gain and inbreeding at a specific region of the genome where a QTL affecting the selected trait is located; (ii) the comparative effectiveness of using marker information combined with pedigree for restricting inbreeding at that position; and (iii) to explore how controlling inbreeding with markers fits into existing theory.
2. Materials and methods
(i) Genetic and population models
A trait under the genetic control of a single known additive bi-allelic QTL and polygenes was simulated. The total genetic value for the ith individual was G i=v i+u i, where v i is the QTL effect and u i is the polygenic effect. The genotypic values due to the QTL for individuals with genotypes BB, Bb and bb were respectively a, 0 and −a, where a is defined as half the difference between the two homozygotes (Falconer & MacKay, Reference Falconer and Mackay1996). The additive genetic variance contributed by the QTL in the base generation (t=0) was thus σq2=2p(1−p)a 2, where p is the initial frequency of the favourable allele B (Falconer & MacKay, Reference Falconer and Mackay1996), which was equal to 0·15.
The QTL was located 50 cM from one end of a 250 cM long continuous segment of the genome. Across the segment, m markers (markers M1), spaced evenly at 10 cM intervals from positions 0 to 250 cM were simulated. At t=0, the number of alleles per marker was 10 for all M1 markers. Marker alleles for a particular individual born at t=0 were sampled from a uniform distribution with an equal probability given to all allelic variants. The M1 markers were used to compute the numerator relationship matrices conditional on pedigree and markers to be used in the optimized selection. In addition, another set of markers (markers M2) was simulated at 1 cM intervals and was used to estimate the observed inbreeding coefficients calculated as the homozygosity by descent, at each position along the genome segment. For each of these M2 markers, all N unrelated individuals (N/2 males and N/2 females) at t=0 were sequentially allocated two unique alleles, i.e., 2N alleles in total were allocated to each M2 marker. Information from these markers was not utilized in the optimized selection procedure. All M1 markers were simulated in linkage equilibrium with each other and with the QTL.
Selection was carried out for eight discrete generations with N offspring produced in each generation providing N/2 male and N/2 female candidates for selection as parents of the next generation. At t=1, the polygenic effect of each individual was sampled from a normal distribution with mean zero and variance σ2u. Alleles at the QTL were randomly allocated according to the starting QTL allelic frequencies. The QTL and polygenes were in linkage equilibrium at t=0. The phenotype of individual j (P j) was obtained by adding the total genetic value (G j) to a normally distributed environmental component with mean zero and variance σ2e. In subsequent generations, the polygenic effect was calculated as the average polygenic effect of the parents plus a random Mendelian term sampled from a normal distribution with mean zero and variance (σ2u/2)[1−(F s+F d)/2], where F s and F d are the pedigree relationship matrix inbreeding coefficients of the sire and dam, respectively. Marker and QTL alleles were inherited from the parent chromosome segments in classical Mendelian fashion, allowing for recombination assuming the Haldane mapping function. It was assumed that all individuals were genotyped for all M1 markers and for the QTL.
(ii) Estimation of breeding values
The effect of the QTL was assumed to be known without error. The total estimated breeding value was the polygenic estimated breeding value plus the assumed known QTL breeding value. Polygenic estimated breeding values were obtained from best linear unbiased prediction (BLUP) using phenotypic data corrected for the QTL effect (P i−v i), and the polygenic heritability. The breeding value for the QTL was calculated as 2(1−p)a, (1−2p)a and −2pa for individuals with genotype BB, Bb and bb, respectively (Falconer & MacKay, Reference Falconer and Mackay1996), and p was updated at each generation.
(iii) Selection and mating
The optimization algorithm, described by Meuwissen (Reference Meuwissen1997), for obtaining maximum genetic gain whilst constraining ΔF to a specified level was used (Appendix A). The A matrix utilized in the optimization to constrain ΔF was either the numerator relationship matrix conditional on pedigree (Ap) or the numerator relationship matrix conditional on pedigree and markers (Am). The Am matrix was calculated at the position of the genome where there was an intention to restrict ΔF for a particular scenario. The deterministic method of Pong-Wong et al. (Reference Pong-Wong, George, Woolliams and Haley2001) was used to compute Am. Matrix Am becomes the same as Ap when the flanking markers around a genome position become uninformative. Mating of parents resulting from the optimized selection procedure was at random.
(iv) Genetic gain and inbreeding
Average polygenic, QTL and total true breeding values and inbreeding coefficient of individuals born at each generation were computed. The average inbreeding coefficient (F) was calculated as:
where A ii refers to the diagonal element of the numerator relationship matrix. The rate of inbreeding (ΔF) at generation t was calculated as (F t−F t−1)/(1−F t−1). Both F and ΔF were obtained using three different relationship matrices: Ap (F P), Am (F M1) and An (F M2), the numerator relationship matrix conditional on M2 markers. Matrix An was calculated at all positions on the genome where M2 markers were simulated. Elements of An for a particular position were obtained using:
where h x(i),y(j) is 1 if allele x of individual i is identical to allele y of individual j and 0 otherwise. Alleles x and y are either paternally (r) or maternally (m) inherited. The F M2 and ΔF M2 calculated using the An matrix are considered the observed inbreeding coefficient and rate of inbreeding determined by homozygosity by descent for that position on the genome. Each founder received a unique pair of alleles at each locus and therefore identical M2 alleles can only be identical by descent.
(v) Coancestry
Different coancestry parameters were calculated in order to gain insight into the mechanism by which the optimization tool restricts ΔF (Appendix B). These included the calculation of the group coancestry weighted by the candidate contribution solutions of the optimized selection algorithm, the group coancestry based on equal contributions of candidates and the average pairwise coancestry between the males and females weighted by the contribution solutions of the optimized selection algorithm.
(vi) Scenarios investigated
Six scenarios were simulated. In all scenarios, σ2u was 0·2, σ2e was 0·8 and N was 120. Scenario 1 was used to investigate the effect that optimized selection using Ap has on the inbreeding in a genome segment where there are no QTL affecting the trait under selection. In scenario 2, Ap was again used but a QTL with effect a=0·1 on the trait was placed on the genome segment. Scenarios 3–5 were used to investigate the effectiveness of Am constructed 5, 55 and 105 cM away from the QTL locus for constraining inbreeding in the region around the QTL that has effect a=0·1. Scenario 6 was the same as scenario 3 except that the QTL effect was set to zero in order to examine the effect of the restriction achieved in the absence of a QTL under selection. The constraint applied to the desired rate of inbreeding was to 0·02 across all scenarios.
3. Results
When using Am in the optimization, approximately half of the replicates failed to give a solution for all eight generations of selection due to the constraint C not being met (λ0 was negative). Only those replicates where a solution was successfully achieved (positive λ0) in all generations contributed to the results presented. Results for the successful generations of those replicates that did not achieve all eight generations were the same as those observed in replicates that successfully achieved eight generations (results not shown).
(i) Genetic gain and QTL allele frequency
Polygenic (G P), QTL (G Q) and total (G T) genetic gains and frequency of the favourable QTL allele (p) for scenarios 2–5 are shown in Table 1. Results for scenario 1 are not comparable with those from the other scenarios (the QTL effect was zero in scenario 1, making the overall genetic variation lower for this scenario).
Table 1. Total (GT), polygenic (GP) and QTL (GQ) genetic means and frequency of the favourable QTL allele (p) at generations four and eight under different scenarios with QTL effect (a) and varying the numerator relationship matrix used in the optimization procedure (A) to constrain the rate of inbreeding. The QTL was at position 50 cM

a Measured in genetic standard deviations. Standard error: from 0·005 to 0·015.
When the QTL had an effect on the selected trait and Ap was used in the optimization (scenario 2), p increased from 0·15 at t=0 to 0·94 at t=8. In contrast, when Am calculated at 5 cM from the QTL position was used (scenario 3), p rose to only 0·61 in generation 8. However, G T was higher in scenario 3 compared with scenario 2, with lower G Q being overcompensated by a higher G P.
Results in terms of p and G Q from scenarios where the Am used in the optimization was calculated at 55 and 105 cM from the QTL (scenarios 4 and 5) were very similar to those when using Ap, where no constraint was specifically applied to the QTL region (scenario 2). However, G P was higher in scenarios 4 and 5 than in scenario 2, resulting in a 4 and 3% greater G T in scenarios 4 and 5, respectively.
(ii) Rate of inbreeding
It can be seen that for scenario 2 (Fig. 1 b) the use of Ap to restrict the rate of inbreeding across the genome did not restrict inbreeding in the region around the QTL under positive selection. This is a demonstration of the hitch-hiking effect (Maynard-Smith & Haigh, Reference Maynard-Smith and Haigh1974), which results in a signature selective sweep in which neutral loci close to a QTL under selection change in frequency, resulting in reduced variation in that region. When Am constructed 5 cM from the QTL was used to restrict the rate of inbreeding, the effect of the selective sweep was eliminated (Fig. 1 c). However, when Am constructed 55 cM from the QTL position was used in the restriction (Fig. 1 d), the effect of the selective sweep led to an asymmetric pattern of restriction with the inbreeding coefficients rising more rapidly at positions away from the restriction position adjacent to the QTL. The observed average rate of inbreeding (ΔF M2) achieved in scenarios 1–6 is shown in Table 2. The rate of inbreeding computed from the pedigree (ΔF p) is also shown for comparison. In scenario 1, no QTL effect was simulated and Ap was used in the optimization and, as expected, both ΔF M2 and ΔF P were restricted to the desired value (0·02). However, in scenario 2, when the QTL had an effect on the selected trait, Ap no longer constrained ΔF M2 at the QTL position to the desired value and ΔF M2 at that position rose by 50% relative to scenario 1.

Fig. 1. Observed inbreeding coefficient (F M2) computed from An calculated as homozygosity by descent, at different positions along the genome segment at generations 2 (◆), 4 (■), 6 (▲) and 8 (×) when optimized selection was practised under different scenarios varying in the QTL effect (a) and the relationship matrix used in the optimization procedure (A). The average inbreeding coefficient calculated using Ap (ped) is also shown for comparison.
Table 2. Observed rate of inbreeding (ΔFM2) calculated as homozygosity by descent using An at different positions, and pedigree rate of inbreeding computed from Ap (ΔFP), when different numerator relationship matrices (A) were used in the optimization to constrain the rate of inbreeding. The optimization aimed to restrict ΔF to 0·02. Values in boldface indicate ΔFM2 at the position where the constraint was targeted. The QTL was at position 50 cM with additive effect (a)

a Averaged across generations 3–8. Standard error for scenarios 1 and 2: ~0·001. Standard error for scenarios 3–6: ~0·002.
In scenario 3, where the Am calculated 5 cM from the QTL position was used in the optimization, ΔF P rose to 0·039, nearly 2·5 times greater than the desired value (Table 2). However, the observed ΔF M2 at the QTL position and at the position where the Am imposed the restriction were approximately half the desired value. At positions further from the position of restriction, the observed F M2 in this scenario rose and reached the value of the F P approximately 80 cM from the restriction position (results not shown). The asymptotic value of F P and F M2 (observed at positions away from the QTL) was higher than those seen in scenario 2.
In scenarios 4 and 5, the ΔF P was similar to that observed in scenario 3 and the observed ΔF M2 at the position of restriction in scenarios 4 and 5 (55 and 105 cM from the QTL, respectively) was also approximately half the desired value as in scenario 3 (Table 2). At the QTL position, the observed ΔF M2 was higher than that seen in scenario 3 and also higher than that seen in scenario 2 where Ap was used and no specific restriction was imposed at the QTL region.
(iii) Coancestry
The previous section showed a clear discrepancy between the observed and expected ΔF M2 values at the position of restriction. In order to investigate further the lower than expected observed ΔF M2 when Am was used in the optimization, additional parameters were calculated: (i) the average group coancestry weighted by the optimal contributions (f o); (ii) the average group coancestry assuming that all candidates have equal contributions (f e); and (iii) the average pairwise coancestry between the male and female selected parents weighted by the optimal contributions, i.e., excluding self and within-sex coancestries (f −o) at a given generation. Results are shown for these parameters in Table 3 for two scenarios: scenario 1, where the QTL had no effect on the trait and the matrix used in the optimization was the Ap matrix, and scenario 6, where the QTL had no effect and the matrix used in the optimization was Am calculated at 55 cM.
Table 3. Coancestry, inbreeding coefficientsFootnote a and rate of inbreeding computed from Ap (pedigree) or An (observed) over generations (t) when Ap was used in the optimization (scenario 1) and when Am constructed at 55 cM was used in the optimization (scenario 6). The QTL had no effect on the trait. Last column shows desired constraint imposed on coancestry (C) in optimization in each generation

a f o: average group coancestry weighted by the optimal contributions from eqn (B1); f e: average group coancestry assuming that all candidates have equal contributions from eqn (B2); f −o: average pairwise coancestry between selected male and female parents from eqn (B3); F: average inbreeding coefficient from eqn (1); ΔF is the rate of inbreeding.
As expected, when Ap was used in the optimization (scenario 1), the constraint on ΔF P was achieved at each generation and the values of observed coancestry and inbreeding coefficients at position 55 cM computed from An, and at all other positions along the genome segment computed from An (results not shown), were very similar to those computed from the pedigree. The specific constraint applied was achieved (f o=C) at each generation and f o at t was equal to f e at t+1. The parents selected from the group of candidates had f −o equal to f e and their matings resulted in a group of offspring with F equal to f −o of their parents (i.e., F at t was equal to f −o at t−1). This clearly shows that when Ap is used in the optimization in a situation where there is no QTL, then the desired constraint is achieved across the genome.
When Am calculated at 55 cM was used in the optimization (scenario 6), the observed f o at the restriction position achieved the desired value in each generation (f o=C). However, the pedigree f o, which represents the optimally weighted group coancestry across all neutral loci in an infinitesimal model, had an increased value, which relates to the higher ΔF P observed in scenarios 3–6. When Am was used in the optimization, the magnitude of the observed pairwise coancestry between the selected males and females (f −o) calculated using An was less than that seen when Ap was used in the optimization (scenarios 1 and 2). Lower observed f −o when Am was used in the optimization compared to when Ap was used resulted in a lower observed F M2 in the offspring at the position of restriction, leading to a lower observed ΔF M2 than that used to calculate the desired constraint (C). The same effect was seen when Am was used in the coancestry calculation. The coancestries and inbreeding coefficients calculated using Ap in scenario 6, though higher than the observed values in the QTL region using An, followed the expectation, i.e., f −o was of a similar magnitude to f e, the group coancestry, and the average inbreeding coefficient of the offspring was that expected from f −o.
4. Discussion
This study has quantified inbreeding in the region around a QTL under selection when the ΔF was constrained using a numerator relationship matrix either conditional on pedigree (Ap) or conditional on pedigree and markers (Am) constructed at different positions on the genome. The constraint on ΔF was achieved using optimized selection that constrained the level of group coancestry amongst the candidates selected as parents weighted by the contributions each makes to the next generation.
It has been shown that Am constructed at a specific position on the genome can be used to constrain the level of weighted group coancestry (f o) at that position within the optimization to the desired level (C). This resulted in the desired rate of increase in unweighted group coancestry (Δf e) at that position on the genome. However, the consequent rate of inbreeding measured using the inbreeding coefficients, at the same position, did not follow our expectation as determined by C. The ΔF M2 measured by the observed inbreeding coefficients (F M2) at the restriction position was consistently below our expectation, our expectation being that the rate of inbreeding would follow the rate of coancestry across generations. At positions away from the restriction, the observed ΔF M2 was seen to rise and reach a maximum equal to the unconstrained rate of inbreeding observed in the rate of pedigree inbreeding (ΔF P). The quantitative results of this study were specific to a single QTL allele starting frequency and assumed marker information content; however, qualitatively we have no reason to believe why they should not exemplify more general phenomena.
In the simulation of the base population, the QTL and M1 markers were simulated in linkage equilibrium. However, this should not have a direct impact on the schemes simulated where the QTL is assumed to be known and the marker information is only related to the calculation of Am. By definition, all animals at the base population are assumed to be unrelated regardless of whether they share the same marker allele or not. During the calculation of Am, the marker information is used to determine the inheritance pattern from the parents to offspring, not to determine any further degree of relationship arising before the base generation. However, linkage disequilibrium would have an indirect impact by increasing the need to utilize more distant markers for inferring IBD (Identity By Descent). Hence, the assumption of linkage equilibrium introduces no bias but a small increase in the precision of IBD estimation that could otherwise be achieved by including additional markers.
Under random mating in the absence of self-fertilization, we expect f o(t)≅F t+2 (Caballero & Toro, Reference Caballero and Toro2000), which holds with our expectation from eqn (7) where f e is equivalent to f o as no constraint on group coancestry was assumed. Under pedigree relationship, the use of Ap in optimized selection constrains f o to a desired level, which is then expected to produce an equal restriction on ΔF. This relationship between f o and ΔF was shown to hold true in this and previous studies (e.g., Grundy et al., Reference Grundy, Villanueva and Woolliams1998).
When Am is constructed at a single position on the genome and used in the optimization, as a mechanism to restrict ΔF at that position to a predefined level, the observed ΔF M2 was found to be lower than expected. The theory behind the expectation that f o(t)=F t+2 is dependent on the average frequency of the alleles in the selected male parents being the same as those in the selected female parents. If due to sampling or other effects the gene frequencies of the two sexes of parents selected were different, then we would expect an excess of heterozygous individuals in comparison to our expectation under Hardy–Weinberg equilibrium (Robertson, Reference Robertson1965). If our hypothesis is that such a situation arose when Am was used in the optimization, then we would expect that f o>f −o and a ΔF lower than expected. This was tested using a single replicate of the scenario 3 simulation. In the simulation, Am calculated at 5 cM away from the QTL was used to constrain the group coancestry in the optimization. The gene frequencies of the M2 markers at the position of restriction for the male and female parents weighted by their relative contributions and the expected excess of heterozygotes were calculated following Robertson (Reference Robertson1965) as shown in Appendix C. The results of this exercise demonstrated that the difference between f o and f −o was equal to the expected deviation from heterozygosity (Table 4). This explains why f o was not subsequently realized in F, as F t=f −o(t−1).
Table 4. Demonstration that the deviation of observed average pairwise coancestry between selected male and female parents (f −o) from the observed average group coancestry weighted by the optimal contributions (fo) calculated using An at 55 cM, can be attributed to the excess of heterozygotes created due to the non-random segregation of alleles between sexes. Results are from eight generations (t) of a single replicate of the scenario 3, where Am at 55 cM was used in the optimization

In the previous illustration, an excess of heterozygotes due to variation in sample gene frequency between the parent sexes was demonstrated, which in turn resulted in F being lower than we would expect from f o. There is, however, no reason to expect this pattern of variation in the sampling of alleles between the two sexes of selected candidates. In fact, the sex of the candidates was not a factor in the non-random sampling of alleles. This was tested by randomizing the sex of the selected candidates following selection subject to there being equal numbers of gametes contributed from male and female candidates. Following randomization, the same outcome was obtained as seen in Table 4, with the expected deviation from heterozygosity again being equal to the difference between f o and f −o. This demonstrates that sex does not have a bearing on the non-random sampling of alleles; rather, there is a between-selected-individual non-random sampling of alleles, which results in an excess of heterozygous offspring.
We can explain the repeated excess of heterozygous offspring observed in the following way. The optimization maximizes gain at a given rate of inbreeding. In the scenarios where Am was used in the restriction rather than Ap, fewer individuals were selected with higher average individual contributions, potentially increasing intensity. This potential increase in intensity is facilitated by the nature of Am, which has more variable diagonals and many more small off-diagonal elements than Ap, which estimates average neutral relationship derived from probabilities. The N(N−1) off-diagonal values drive the constraint in the optimization and it is therefore possible to select individuals who have a high proportion of very low or zero relationships with other candidates, and consequently a greater proportion of the group coancestry may arise from self-coancestries, allowing fewer but greater contributions. Such solutions are attractive since it allows greater contributions from individuals with greater breeding values. Therefore, in each generation, the more dispersed values of Am lead to selection of individuals that have low or zero relationships because they are homozygous for alternative alleles, resulting in a higher heterozygosity amongst the offspring than would be expected from the array of candidate genotypes. For the reason that more loci are averaged to obtain the Am used, the deviation from the expectations of Ap diminish as the dispersion among the elements of Am diminish (T. Roughsedge, unpublished results).
Having established that the discrepancy between f o and f −o was due to non-random inheritance of alleles at the position of restriction in the selected candidates, we can hypothesize that removing the constraint at t−1 and allowing random selection to take place would restore the expected relationship between group coancestry and average inbreeding at t. In order to test this hypothesis, a further simulation was run. The scenario 3 simulation parameters were again used, but for generation 7 random selection rather than optimized selection was practised, with 20 of the candidates of each sex randomly selected as equally contributing parents. The simulation was run for 1000 replicates. The optimization makes six generations of selection with the constraint on the rate of inbreeding being applied using Am. If the discrepancy between f o and the subsequently realized F M2 was due to the selection of parents with non-random samples of alleles, then the introduction of random selection should select a sample of parents with a random sample of the alleles from the previous generations parents, which were selected under the optimization. In the simulation, the F M2 realized in t=8 was approximately equal to the f o at t=6 (Table 5). This further demonstrates that the optimized selection method constrains group coancestry to the desired level, but non-random segregation of allele frequency at the restriction position occurs in selected parents. The discrepancy between f o and the realized ΔF M2 can be attributed to this non-random segregation of alleles between the selected parents.
Table 5. Effect on coancestry and inbreeding calculated using An at 55 cM over generations (t), when introducing random selection at t=7 and t=8 following optimized selection using Am (55 cM) at t=1–6

a f o: average group coancestry weighted by the optimal contributions from eqn (B1); f e: average group coancestry assuming that all candidates have equal contributions from eqn (B2); f −o: average pairwise coancestry between selected male and female parents from eqn (B3); F: average inbreeding coefficient from eqn (1).
b Desired constraint used in optimization. Applied at t=1–6 in which optimized selection is undertaken.
This raises a question about which rate of inbreeding it is appropriate to constrain when undertaking selection. In the optimized selection procedure, we actually constrain the rate of group coancestry:
By constraining the rate of group coancestry, the loss of genetic variation is managed but no absolute control is placed on the inbreeding coefficients. As we have seen in this study, when relationships are constructed at a single locus and the group coancestry at this locus is constrained, the relationship between group coancestry at t and average inbreeding coefficients at t+2 breaks down. The loss of genetic variation is still managed but ΔF is less than we would expect. This highlights why we should treat the management of genetic variation and inbreeding coefficients as separate issues.
A technique to simultaneously constrain both pedigree group coancestry and also group coancestry around a QTL under selection may provide a solution to simultaneously being able to constrain the ΔF M2 on the segment of the genome around the QTL and the pedigree ΔF P. This would require an approach that allowed two or more constraints to be introduced to the quadratic optimization such as semi-definite programming (Pong-Wong & Woolliams, Reference Pong-Wong and Woolliams2007). Such an approach could be useful when a QTL is segregating in a region where a loss in genetic variability is not desirable. Here, a constraint on both the group coancestry in the region around the QTL and also the pedigree group coancestry could avoid a loss in genetic variation in the region and also the average genetic variation.This study has shown that it is possible to erode the relationship between the rate of coancestry and the rate of inbreeding under random mating. Hence, we have reinforced the concept that in order to manage the loss of long-term genetic variation, group coancestry should be managed rather than the inbreeding coefficient. If, however, there is interest in the effects of inbreeding depression, then the management of group coancestry based on a numerator relationship matrix conditional on pedigree and marker information does not provide a reliable estimate of inbreeding coefficients. In order to manage inbreeding depression, it is advisable to take a two-stage approach with the first stage managing the loss of variation through the constraint of group coancestry, followed by mate allocation to manage inbreeding depression. This message has a clear bearing on both selection and conservation breeding programmes.
We are grateful to the SEERAD for funding this work. R. P.-W. and J. A. W. acknowledge funding from the BBSRC.
Appendix A. Optimized selection
The optimization algorithm, described by Meuwissen (Reference Meuwissen1997), maximizes the objective function:
where ct is the solution vector (of dimension N) of optimal contributions of candidates at generation t, g is the vector of total estimated breeding values, A is the numerator relationship matrix N×N of candidates, Q is a known incidence matrix N×2 with ones for males and zeros for females in the first column and ones for females and zeros for males in the second column, C is the constraint on the rate of inbreeding, ½ is a vector of halves of dimension 2, and λ0 and λ (a vector of dimension 2) are Lagrangian multipliers. The Lagrangian multiplier λ0 is chosen such that ctTAct/2 is constrained to 1−(1−ΔF)t (Grundy et al., Reference Grundy, Villanueva and Woolliams1998), where ΔF is the desired rate of inbreeding.
Appendix B. Coancestry formulae
All three relationship matrices (Ap, Am, An) can be decomposed into four sub-matrices, Amm, Amf, Afm and Aff, each of dimensions N/2×N/2, relating to the male by male, male by female, female by male and female by female numerator relationships, respectively. The solution vector of optimal parental contributions (c) can also be decomposed into two sub-vectors, one for male (cm) and one for female (cf) parent contributions, each of dimensions N/2. The c vector sums to 1 and each sub-vector (cm and cf) sums to 0·5. The average group coancestry of candidates (f o) calculated using their optimal contributions is the average pairwise coancestry of a given group of individuals including within-sex and self-coancestries weighted by their contributions, and is obtained from the optimization procedure as:
This differs from the group coancestry (f e) defined by Cockerham (Reference Cockerham1967) in which all candidates are assumed to make an equal contribution. Let ce be a vector of dimension N with all elements equal to 1/N (i.e., all candidates make an equal contribution). Then, the equal contributions group coancestry (f e) can be calculated as
Finally, the average pairwise coancestry between the male and female individuals selected as parents weighted by their contributions (f −o) is:
where 0 is a vector of dimension N/2 with all elements set to zero.
Appendix C. Excess of heterozygotes when allele frequency differs between parents
Let q 1 be the frequency of allele A of a bi-allelic gene in a group of male parents and q 2 be the frequency in a group of female parents. Then, following random mating, the expected proportion of heterozygotes in the offspring is q 1(1−q 2)+q 2(1−q 1). However, the mean gene frequency of the parents is (q 1+q 2)/2, and from the Hardy–Weinberg equilibrium we would expect the frequency of heterozygotes in the offspring to be (q 1+q 2)(1−(q 1+q 2)/2). Therefore the excess of heterozygotes when q 1≠q 2 is ½(q 1−q 2). This theory was extended beyond the bi-allelic case by Robertson (Reference Robertson1965) :
where H is the excess of heterozygotes, q x,y is the frequency of allele x in sex y and L is the total number of possible alleles at the locus considered.
The deviation of the observed f −o from the observed f o at t calculated using An at the restriction position when Am is used in the optimization (e.g., Table 3, scenario 6) is accounted for by the deviation in expected heterozygotes calculated in eqn (A4). This results in the following equality:
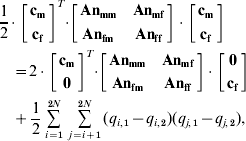
where Anxy is the sub-matrix between sexes x and y of the numerator genetic relationship matrix calculated at the same position as Am was constructed; cx is the contributions vector for sex x; q i,1 is the frequency of allele i in sex 1, q i,2 is the frequency of allele j in sex 2, q j,1 is the frequency of allele j in sex 1 and q j,2 is the frequency of allele j in sex 2, 2N is the total number of M2 alleles.






