


Motor status epilepticus can be generalized or focal. The latter is known as epilepsia partialis continua (EPC) or Kojevnikov’s epilepsia.Reference Valentin, Ughratdar, Cheserem, Morris, Selway and Alarcon 1 EPC is a rare seizure type characterized by rhythmic or semi-rhythmic clonic or myoclonic jerks localized to a unilateral focal region. It is notoriously difficult to treat and may persist for from hours to years.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2 We present a case of EPC secondary to focal cortical dysplasia (FCD) lasting for 50 years.
A 50-year-old, right-handed, otherwise healthy male was assessed in our comprehensive epilepsy clinic for ongoing seizures. He has continuous myoclonic jerks affecting the left hemibody (video available online; see Supplementary Materials), consistent with EPC, and focal seizures with loss of awareness, which are controlled by the antiseizure medications phenytoin 375 mg daily and primidone 1,000 mg daily. He stopped consulting physicians following unsuccessful resective surgery at age 17 years prior to our evaluation, during which no treatment modifications were made.
The seizures began on day six of life. These included EPC as well as discrete focal seizures characterized by left arm numbness progressing to involve the whole upper limb, then clonic jerking of the left hemibody and postictal left Todd’s paresis lasting 10–15 minutes. Loss of awareness occurs occasionally and rarely evolves into bilaterally synchronous tonic–clonic seizures. These events are well-controlled on medications. The last event occurred five years ago.
Developmental milestones were delayed: sitting at 12 months, walking at 18 months, and speaking in sentences at 5 years. He attended special education classes. There were no additional seizure risk factors.
Physical examination revealed mild left-sided weakness of the face, arm, and leg. Fine finger movements were decreased on the left. Deep tendon reflexes were symmetric, and there was a left extensor plantar response. There were no sensory deficits.
The patient was previously evaluated in the epilepsy monitoring unit at age 17 years, when he had frequent episodes of status epilepticus with loss of awareness that required hospital admissions. EEG demonstrated nearly continuous right central-sagittal-parietal spikes. Neuropsychological assessment revealed an IQ of 66, left-sided sensorimotor deficits, and likely left hemispheric language dominance. He underwent right-sided craniotomy with electrocorticography (ECoG). Initial inspection of the brain revealed an extremely small gyrus at the upper precentral gyrus and a very small postcentral gyrus (Figure 1). Nearly continuous ECoG discharges occurred in the right parietal lobule, postcentral gyrus, lower precentral gyrus, and frontal operculum. The epileptogenic region extended over the Rolandic cortex. The right superior parietal lobule was resected. Postresection ECoG demonstrated abundant discharges over the post- and precentral gyri, but these were not resected due to possible functional compromise.
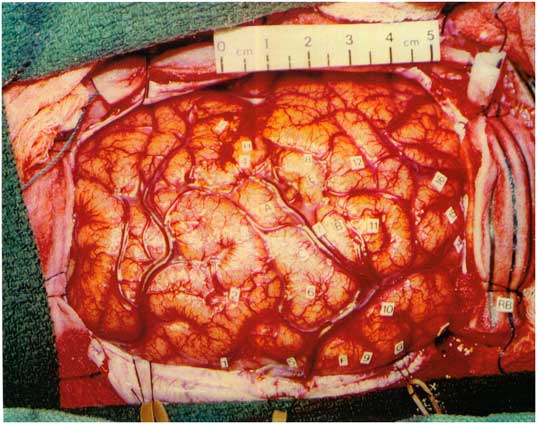
Figure 1 Intraoperative photo prior to resection. Most active spike foci were right parietal lobule behind the postcentral sulcus (11), right postcentral gyrus (10), right lower precentral gyrus (7), and right frontal operculum (4).
Neuropathology revealed transcortical gliosis and Chaslin’s subpial change (Figure 2A,B). Neocortical lamination ranged from near normal to markedly disrupted (Figure 2C). Dysmorphic neurons (hypertrophic, pleomorphic, prominent nucleoli, coarse Nissl substance) were present through all layers of cortex and subcortical white matter, but were most numerous deep to layer II in regions of cortex with the greatest degree of laminar disorganization. Dysmorphic neurons revealed frequent clustering and random apical polarity (Figure 2D–H). Pathology was reanalyzed in 2016 with immunohistochemistry, revealing select dysmorphic neurons, expressed phosphorylated high-molecular-weight neurofilaments, and/or abundant synaptophysin in the perinuclear soma (Figure 2F). These findings are consistent with ILAE type IIa cortical dysplasia,Reference Blumcke, Thom and Aronica 3 a classification and diagnosis not available at the time of surgery.
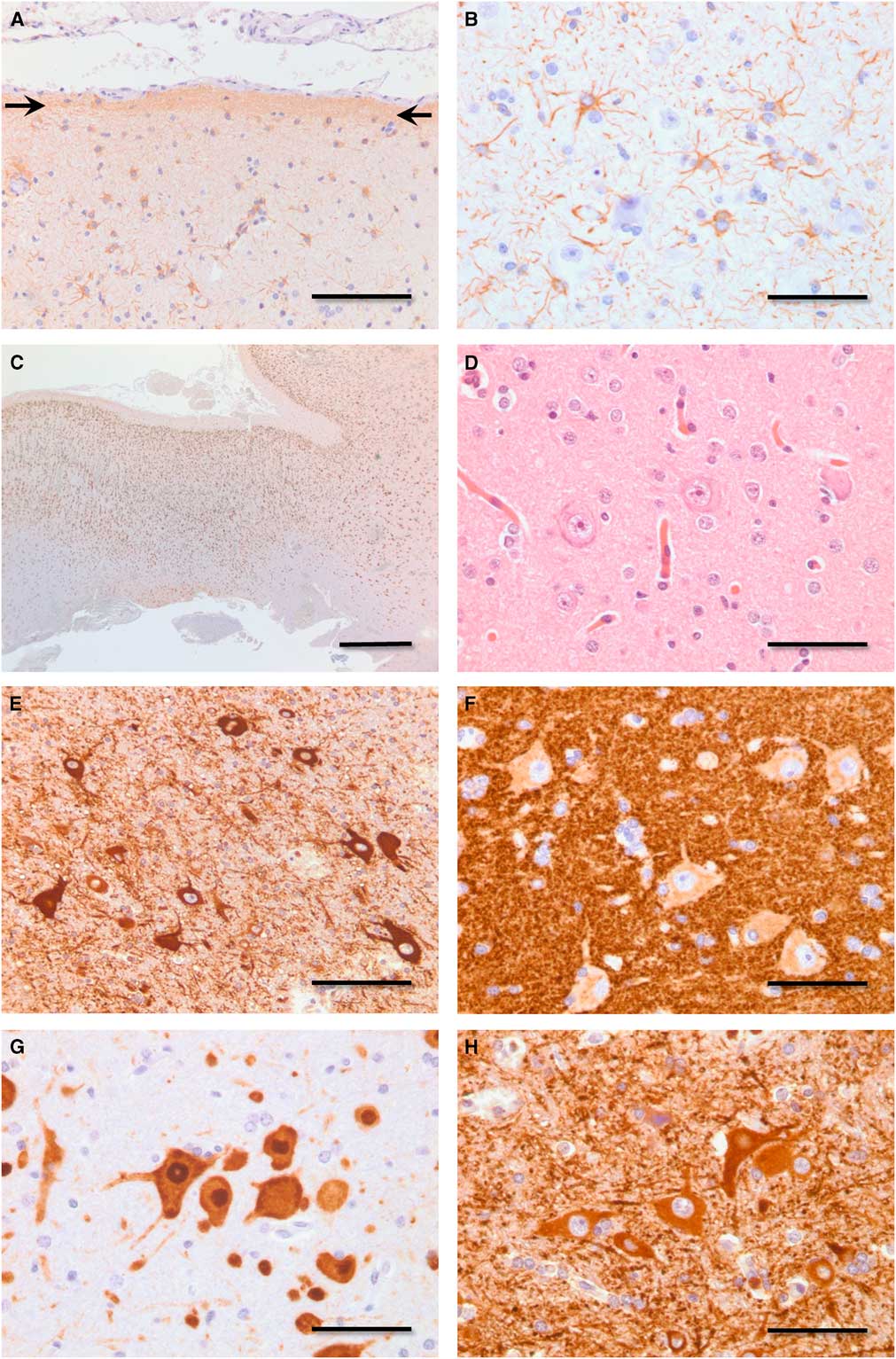
Figure 2 Photomicrographs demonstrating pathology. (A) Chaslin’s subpial gliosis (arrows, anti-GFAP immunoperoxidase, bar=100 μm). (B) Transcortical gliosis (anti-GFAP immunoperoxidase, bar=50 μm). (C) Disrupted neocortical lamination. Moving from left to right, the neocortex in this field of view ranges from near normal to one of marked laminar blurring (anti-NeuN immunoperoxidase, bar=1 mm). (D) Dysmorphic neurons in various neocortical laminae display hypertrophy, nuclear enlargement, and nucleolar prominence (hematoxylin and eosin, bar=50 μm). (E) Dysmorphic neurons demonstrate lack of common polarity (anti-MAP2 immunoperoxidase, bar=100 μm). (F) Dysmorphic neurons reveal random polarity and increased expression of synaptophysin in the perinuclear soma (anti-synaptophysin immunoperoxidase, bar=50 μm). (G) Dysmorphic neurons demonstrate lack of regular size, spacing, and polarity (anti-NeuN immunoperoxidase, bar=50 μm). (H) Dysmorphic neurons reveal random polarity, pleomorphism, and clustering (anti-MAP2 immnoperoxidase, bar=50 μm). GFAP=glial fibrillary acidic protein; NeuN=neuronal nuclear antigen; MAP2=microtubule-associated protein.
Following surgery, seizures became controlled with antiseizure medications; however, the continuous myoclonic movements persisted. His EEG showed nearly continuous spikes over the right fronto-central-sagittal region.
EPC is challenging to diagnose and manage. Two groups of EPC have been recognized with different etiologies and disease trajectories. Type I is usually nonprogressive, associated with a Rolandic lesion, stable focal deficit, and normal neuropsychological profile, whereas type II typically has a progressive course with additional neurological signs, sleep, and behavior disorders, progressive mental deterioration, and a characteristic EEG with long subclinical paroxysms of slow spikes.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2 Type I EPC is associated with vascular lesions, tumors, and other static lesions, and type II with encephalitides, particularly Rasmussen’s encephalitis.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2 Focal cortical dysplasias (FCDs) have also been associated with EPC.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2
By definition, EPC lasts at least for an hour, but it can last for years.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2 However, most EPC lasts for from days to weeks. The longest durations of EPC are reported with FCDs.Reference Kravljanac, Djuric, Jovic, Djordjevic, Zamurovic and Pekmezovic 2 No previous case has reported the neuropathology associated with prolonged EPC. This case demonstrates the pathology of FCD associated with this patient’s EPC. As the most active region of epileptogenicity found on ECoG was over the Rolandic cortex and not resected, it is likely responsible for the ongoing EPC.
The pathophysiology behind EPC is poorly understood, with both cortical and subcortical influences described.Reference Guerrini 4 Cortical abnormalities in the Rolandic cortex underlie many of these lesions. However, even in their absence, fast rhythmic bursting has been seen in patients with EPC or fast cortical tremors.Reference Guerrini 4 Why the motor cortex is so sensitive to these abnormalities is unclear. Disruption of a thalamocortical loop has been shown to result in EPC, and the basal ganglia may also have a role in generating EPC, possibly through a lack of inhibition.Reference Guerrini 4
Treatment of EPC is notoriously difficult, and epilepsy surgery is not frequently considered due to the risks of functional compromise, as in the case presented herein. However, neurostimulation, either of the thalamus or motor cortex, theoretically may be useful given what is known about the pathophysiology of EPC and the involvement of the motor cortex, as well as the possible role of the thalamus. Indeed, a cortical stimulator has been placed with success in two cases of EPC, resulting in resolution of EPC as well as improvement in self-limited seizures.Reference Valentin, Ughratdar, Cheserem, Morris, Selway and Alarcon 1
We have presented here a case with EPC lasting for the patient’s lifetime of 50 years, likely secondary to FCD given the pathology of the resected tissue. Following epilepsy surgery, the patient was free from disabling seizures with loss of awareness, but the EPC persisted. The pathophysiology of EPC may involve both cortical and subcortical mechanisms. This suggests the difficulties in treating EPC due to location in eloquent cortex that may be improved with neurostimulation.
Consent
Written informed consent was obtained from the patient for publication of this report and the accompanying images. A copy of the written consent is available for review from the editor-in-chief of this journal.
Statement of Authorship
Kristin M. Ikeda was responsible for study concept and design, as well as for editing and writing the report. Mubarak M. Aldosari was responsible for the literature review and writing the report. Huda AlGhefari was responsible for writing the report, creating the pathology images, and working on the text. Robert R. Hammond was responsible for creating the pathology images, editing the report, and supervision. Seyed M. Mirsattari was responsible for overall study concept and design, patient information, editing the report, and supervision.
Disclosures
Kristin M. Ikeda, Mubarak M. Aldosari, Huda AlGhefari, Robert R. Hammond, and Seyed M. Mirsattari hereby declare that they have no conflicts of interest disclose.
Supplementary Material
To view the supplementary material for this article, please visit https://doi.org/10.1017/cjn.2017.237.



