


The differential diagnosis of paroxysmal hyperkinetic movements is extensive. The major categories are movement disorders and seizures (e.g. reflex seizures, tonic seizures in LGI 1 encephalitis); other diagnoses to consider are hyperekplexia, episodic ataxias, sleep disorders, and psychogenic events. A thorough assessment, attention to details of the clinical presentation, response to therapy, and appropriate investigations are required for the correct diagnosis.
We present a patient with longstanding history of paroxysmal events involving mainly her right arm, whose diagnosis took a long time to be achieved due to the semiological overlap with paroxysmal movement disorders.
An 18-year-old right-handed first year university student was referred to the Comprehensive Epilepsy Centre in London Health Sciences Centre (LHSC) from the Movement Disorders Clinic at LHSC after an 11 year history of brief episodes which consisted of sudden onset jerks of the right arm associated with flexion of the right elbow and the right wrist and abduction of the right shoulder. In some episodes, she would also exhibit head jerks to the right, pulling of the right side of her face causing facial grimacing, and stiffening of her right leg. She occasionally experienced a very brief subjective shaking sensation, without real jerking, prior to the attacks. She retained full awareness during her attacks. They were mainly triggered by voluntary movements, such as jumping and running. Even riding a school bus could trigger them. They occurred more frequently with emotional stress, insomnia, and viral upper respiratory tract or gastrointestinal infections. She had rare unprovoked events, as well. She could have multiple daily episodes, up to 100/day, each lasting between 10–30 seconds. Most of them occurred during wakefulness. She experienced two generalized tonic-clonic seizures within four hours of each other at the age of 13 years, which followed a reduction in the dose of her oxcarbazepine. The first seizure occurred during her sleep at three a.m. while the second one, in the Emergency Department, lasted 1 ½ minutes. These events, occurring shortly after a reduction in her oxcarbazepine dose, were not preceded by the episodes described above or any other warning symptom. Previous electroencephalograms (EEGs), including prolonged video-EEG recordings for a few days at the age of eight years, had reportedly been normal or non-specific. Multiple anatomical magnetic resonance imaging (MRI) studies of the brain were normal. Blood and urine tests including serum lactate, thyroid functions, serum and urine amino acids had failed to find a metabolic or immunological cause. A complete neuropsychological assessment was essentially normal. She was taking lamotrigine 550 mg daily, oxcarbazepine 1500 mg daily, and clobazam 22.5 mg daily. She had previously tried many medications, including levodopa, tetrabenezine, phenytoin, gabapentin, levetiracetam, clonazepam, and acetazolamide, which were either ineffective or caused side effects. Only phenytoin had a mild benefit but it was discontinued prematurely after a mild rash. She has never tried carbamazepine or valproic acid.
Her past medical history included septicemia at the age of 2 ½ years, with full recovery and no seizures at that time. When she was four years old, she had short episodes of dizziness, diplopia, and unsteadiness on a daily basis for four months that did not respond to flunarazine and resolved spontaneously. She had severe migraine attacks with some stabbing features of the pain, unrelated to her spells, between the ages of 12 to 14 years that were treated with indomethacin with some benefits. She was schooled at home by her mother for most of her grades seven to nine because of those headaches. A maternal great uncle (grandmother’s brother) had one seizure in his life, another maternal great uncle (her grandfather’s brother) had seizures since childhood and died of an unknown cause at the age of 40 years. Her father had migraine headaches. Her neurological and general physical examination was unremarkable, including her motor and sensory functions. With the main differential diagnosis of paroxysmal dyskinesia and frontal lobe epilepsy, particularly peri-rolandic seizures, she was admitted to our Epilepsy Monitoring Unit for a prolonged video- EEG telemetry.
During nine days of continuous video-EEG recording, interictal EEG was normal in wakefulness and sleep. More than 30 stereotyped episodes (video sequence) were captured, all of which were associated with left frontal-central epileptic discharges (Figure 1). Her attacks were elicited by running. Ictal minus interictal single-photon emission computed tomography (SPECT) studies did not show significant changes, except for possible subtle hyperperfusion in the mesial frontal region, which could not be appreciated quantitatively. Based on the semiology and EEG findings, we hypothesized that the episodes were epileptic seizures that arose from the left peri-Rolandic areas. Subdural EEG recording was obtained to confirm epilepsy, better define the epileptogenic zone for a possible resective surgery and accurately map function of the cerebral cortex in that area. While waiting to be admitted for invasive EEG recordings, clobazam was replaced by clonazepam 0.5 mg daily and lacosamide 300 mg daily was added, which were ineffective. The subdural map included electrode lines covering bilateral mesial frontal-parietal areas and left frontal-parietal convexity in order to cover the primary sensory and motor cortices, supplementary sensory-motor area (SSMA), and the left premotor cortex. During nine days of invasive EEG recording, 142 episodes were captured, all of which were consistent with epileptic seizures that were localized to the left rolandic and adjacent left premotor cortices and the left SSMA (Figure 2). Cortical stimulation and functional MRI using motor and sensory paradigms were obtained to map function of the involved areas. Several clinical seizures were induced by three to five mA electrical stimulations before and during the surgery. The epileptogenic zone included primary motor cortex for the right hand and arm areas, the left premotor cortex anterior to them, and the left SSMA anterior to the rolandic foot area. Responsive cortical stimulation was considered but the device was not available in Canada. She underwent a partial corticectomy of the epileptogenic area within the left rolandic cortex plus a larger resection in the right premotor and SSMA anterior to the rolandic area of the right foot. No surgical tissue was submitted for pathology. Postoperatively, she developed a very mild right hand and foot weakness that recovered completely with physiotherapy in less than one month. Eighteen months later, she remains seizure-free on lower antiepileptic drugs (AEDs) dose.
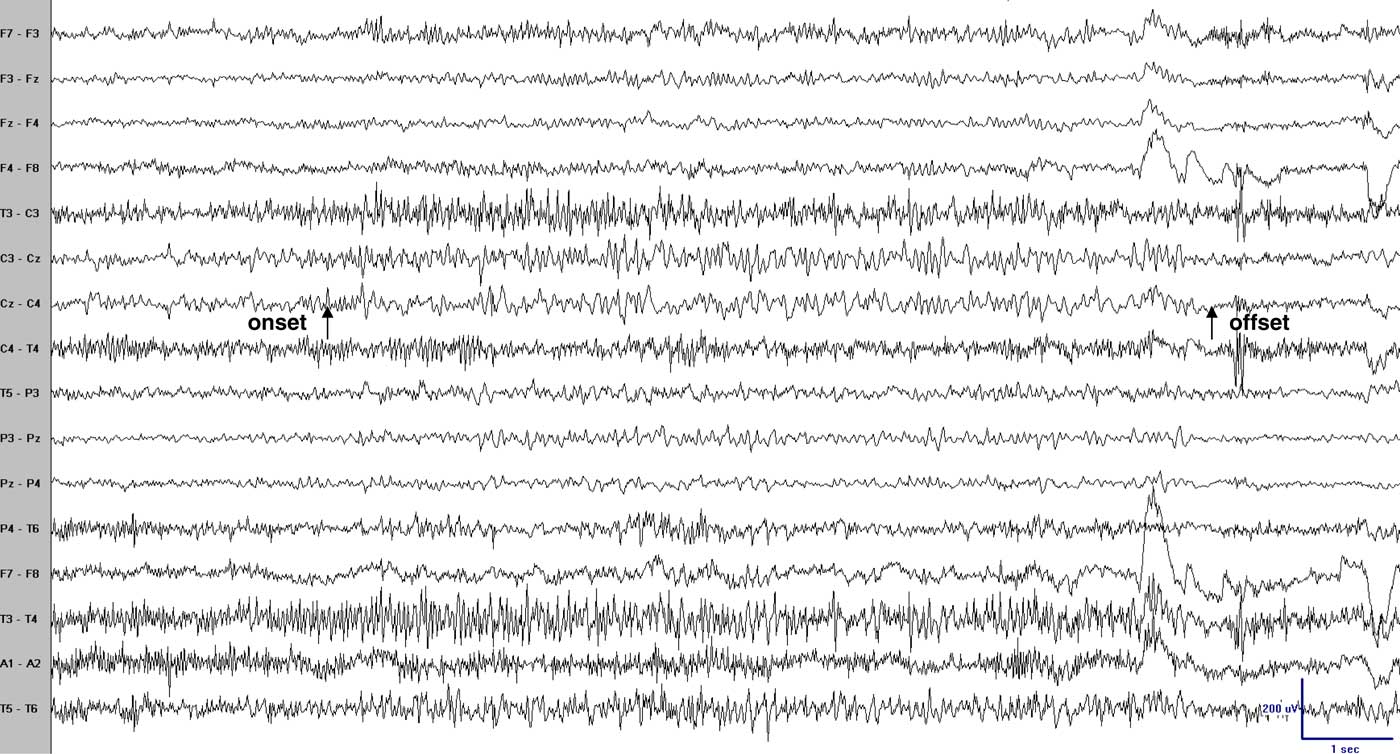
Figure 1 Scalp EEG (coronal montage) during one episode revealed sudden onset, low voltage 30–40 Hz activity principally at electrodes Cz-C3 with spread to P3 and F3, decreasing in frequency and increasing in voltage with an abrupt offset. Arrows point to the clinical onset and offset. Bipolar recording: low frequency filter (LFF)=1 Hz; high frequency filter (HFF)=70 Hz; notch filter off; sensitivity=10 uV/mm.
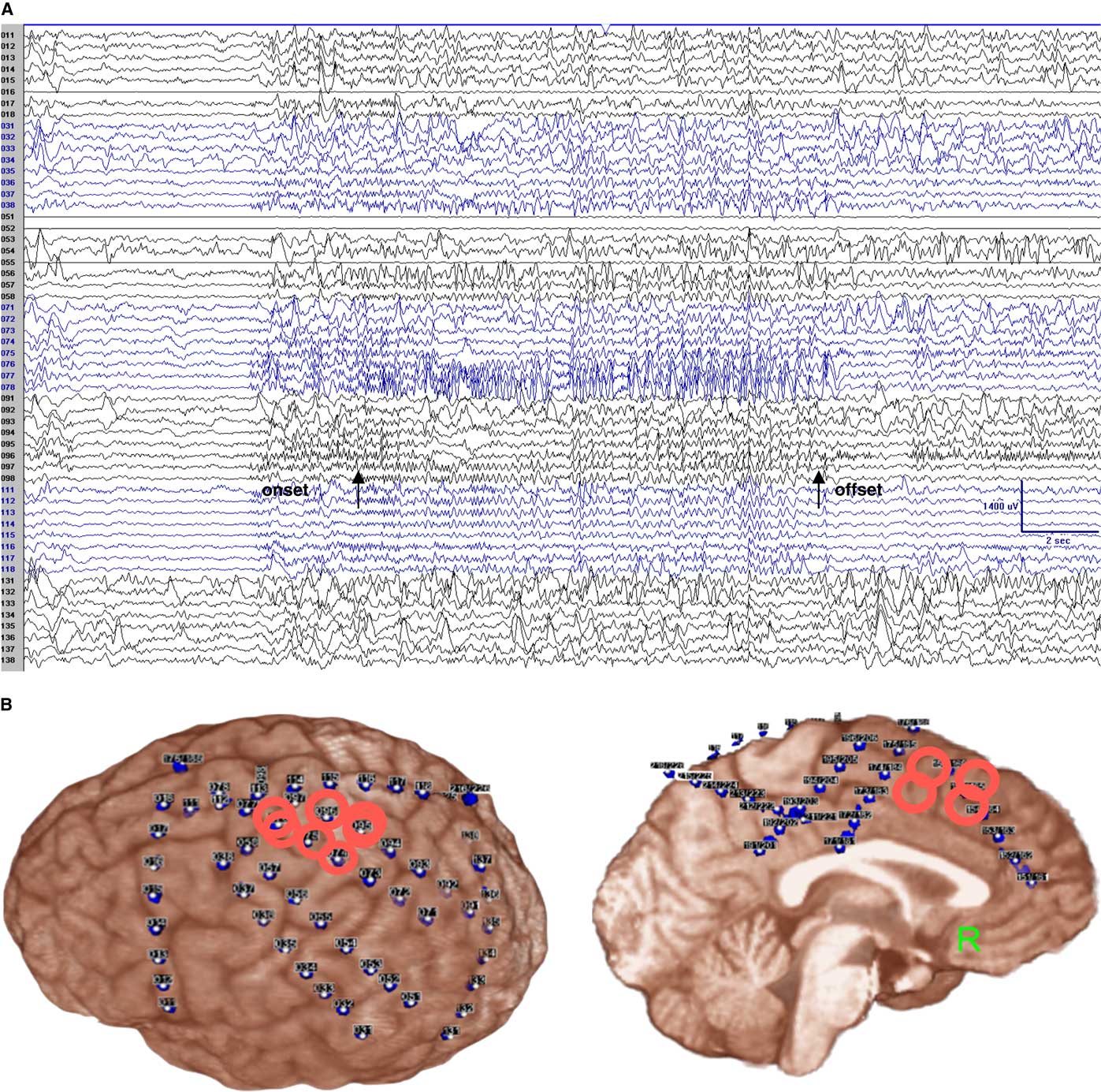
Figure 2 Subdural recording (monopolar montage) showed high frequency and low voltage rhythmic polyspikes mainly over electrodes 76–78 (the rolandic cortex for the right hand and right arm, not resected), with simultaneous involvement of the adjacent electrodes (electrodes 37–38, 55–56, 95–97) plus the adjacent left SSMA (electrodes 174–175, 194–195; not shown here), lasting about 15 seconds. Arrows point to the clinical onset and offset. Referential recording: LFF=1 Hz; HFF=70 Hz; notch filter off; sensitivity=70 uV/mm (A). Subdural EEG coverage shows that the main epileptogenic area (surrounded by red circles) is over the left rolandic cortex (B).
This case illustrates difficulties in the diagnosis of some patients with paroxysmal hyperkinetic movements, namely to distinguish between paroxysmal dyskinesias and reflex epilepsy manifesting with peri-rolandic seizures. Furthermore, they can co-occur in some families, because the two conditions can share a common genetic background.Reference Guerrini, Bonanni and Nardocci 1
Among the recognized subtypes of paroxysmal dyskinesias, the most frequent type is the kinesigenic one (PKD), first described in 1967.Reference Kertesz 2 The current diagnostic criteria for PKD are: the presence of an identified kinesigenic trigger, attack duration less than one minute, no loss of consciousness or pain, normal interictal exam, age at onset between one and 20 years, good control with carbamazepine or phenytoin, and exclusion of other diseases.Reference Bruno, Hallett and Gwinn-Hardy 3 Furthermore, clinical manifestations of PKD are less uniform in children; at this age attacks can occur during sleep.Reference Bruno, Hallett and Gwinn-Hardy 3 On the other hand, peri-rolandic seizures are characterized by clonic movements and twitching of one or more extremities, with involvement mainly of the distal muscles. They can be very frequent, several episodes per day, during wakefulness or sleep, may be preceded by a sensory aura, and can involve the head. Scalp EEG, interictal and ictal, can be normal if the involved brain surface area is small. If it is refractory to AEDs, tailored surgical resection can be a treatment option.Reference Pondal-Sordo, Diosy, Tellez-Zenteno, Girvin and Wiebe 4
In the present case, the spells met most of the diagnostic criteria for PKD. However, there were several “red flags” that led to the diagnosis of reflex epilepsy manifesting with peri-rolandic seizures. First, even though her history of two generalized tonic-clonic seizures within four hours did not meet the diagnostic criteria for epilepsy, they pointed towards an epileptic tendency. Triggers that imply voluntary movements may be unveiling a proprioceptive sensory input to hyperexcitable cortical motor neurons. Second, she had had rare unprovoked episodes during sleep. Third, despite the fact that she never tried carbamazepine, which is the treatment of choice for PKD, she took phenytoin, which is also included in the diagnostic criteria for PKD, without benefits. In addition, she took oxcarbazepine without benefit, which would have been expected to be as effective as carbamazepine due to similar mechanism of action. Fourth, the scalp EEG findings during the episodes were supportive of an epileptic cause that was confirmed with invasive EEG and postoperative outcome. The SPECT imaging did not reveal significant perfusion changes. However, SPECT is not reliable for differentiating seizures and PKD attacks because the latter can show ictal cortical hyperperfusion during the attack, as seizures do.Reference Kim, Kim and Chung 5
In summary, movement disorders and reflex epilepsy may overlap in some patients. A typical example is the misdiagnosis between PKD and peri-rolandic seizures. A correct diagnosis of reflex epilepsy and PKD is crucial given the difference in treatment and prognosis between them. A detailed analysis of the semiology of events and targeted investigations like video-EEG recording along with empiric therapy are necessary for the accurate diagnosis.
Acknowledgements
The authors thank the patient, her family and all people who took care of her during the diagnostic and treatment procedure, particularly the nurses who took care of her during the admissions at the Epilepsy Monitoring Unit.
Disclosures
Asier Gomez-Ibañez, Kiran Khanapure, David Steven, and Seyed Mirsatarri do not have anything to disclose.
Courtney Casserly has the following disclosures: Biogen Idec, Clinical Fellowship, Funding for fellowship at the University of Toronto; EMD Serono, Meeting attendance, Funding for meeting attendance.
Conflicts of Interest
The authors declare that this work has not been supported for any grant or industrial company. This clinical case was presented at the 49th Annual Congress of the Canadian Neurological Sciences Federation in June 2014 and the annual meeting of the American Clinical Neurophysiology Society in February 2015.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi./org/10.1017/cjn.2015.326



