Migraine is a severe neurological disorder that causes an intense throbbing or pulsating pain in one area of the head and can be accompanied by nausea, vomiting and extreme photophobia, and phonophobia (Rose & Capildeo, Reference Rose and Capildeo1983). Migraine is divided into two main categories according to criteria set out by the International Headache Society, namely migraine with aura (MA) and migraine without aura (MO). This disease affects approximately 12% of the Caucasian population, with two-thirds of affected cases being female. Attacks may last up to 72 hours, causing a significant personal burden as well as adversely affecting work productivity. Given the epidemiology of the disease burden, with mainly the economically productive age group being affected, the World Health Organization (WHO) has ranked migraine in the top 20 most debilitating diseases (Leonardi et al., Reference Leonardi, Steiner, Scher and Lipton2005). Despite this significant personal and economic burden of migraine, current treatments are only effective for a proportion of sufferers.
The exact pathophysiology of migraine is still not fully understood and even though a significant proportion of the disease has been attributed to genetic heritability, few causal genetic variants have been linked to common forms of migraine (Schurks, Reference Schurks2012). For human diseases and other complex traits, heritability can be estimated from the concordance rate between monozygotic and dizygotic twins (Macgregor et al., Reference Macgregor, Cornes, Martin and Visscher2006). Migraine has been shown through twin and family studies to have a significantly heritable component, which has driven genetic studies aimed at identifying causal variants (Colson et al., Reference Colson, Lea, Quinlan and Griffiths2006). Heritability estimates for migraine according to population twin studies range between 0.34 and 0.57 (Mulder et al., Reference Mulder, Baal, Gaist, Kallela, Kaprio, Svensson and Palotie2003; Svensson et al., Reference Svensson, Larsson, Waldenlind and Pedersen2003). While Familial Hemiplegic Migraine (FHM) studies and large migraine Genome-Wide Association Studies (GWAS) have significantly contributed toward our understanding of the possible mechanisms involved in migraine pathogenesis, further research identifying association between migraine susceptibility and common genetic variants is needed (Maher & Griffiths, Reference Maher and Griffiths2011).
Current theories pertaining to the mechanistic cause of migraine attacks describe a neurovascular mechanism whereby a range of stimuli activate the trigeminal nervous system, causing pain sensitisation via the thalamus. Other theories place more emphasis on a dysfunction between the neuronal nuclei, which in turn modulates sensory inputs (de Almeida et al., Reference de Almeida, Leao, Gomes, da Silva and Teixeira2009; Varlibas & Erdemoglu, Reference Varlibas and Erdemoglu2009). Neurogenic inflammation was found to be a key mechanism in activating the trigeminal nervous system (Moskowitz, Reference Moskowitz2007). Previously, emphasis has been placed on the idea that vascular and neural stimuli resulting in a wave of neuronal hyperexcitability cause cortical spreading depression (CSD) and consequently a migraine attack. The hypothesis further extends to state that meningeal inflammation occurs as a consequence of CSD and local mediators such as calcitonin gene-related peptide (CGRP), and substance P activates meningeal sensory neurons, causing activation of the pain pathway rather than vasodilation (Buzzi, Reference Buzzi2001). However, while CSD has been well documented to cause MA, there is still debate about the importance of CSD in MO (Lauritzen, Reference Lauritzen1987).
New studies have suggested that migraine pain and trigeminovascular activation are caused by a central mechanism which does not require a primary sensory input (Goadsby & Akerman, Reference Goadsby and Akerman2012; Lambert et al., Reference Lambert, Truong and Zagami2011). The most recent theory describing migraine pathogenesis states that migraine is caused by a dysfunction of the subcortical brain structures including the brainstem and diencephalic nuclei which are involved in modulating sensory inputs. The theory suggests that aura is triggered by dysfunction of these nuclei and that the same mechanism is responsible for the pain and other symptoms experienced during migraine attacks (Akerman et al., Reference Akerman, Holland and Goadsby2011).
CGRP is a vasoactive peptide released during migraine attacks and has also been shown to correlate with peripheral trigeminal activation. Trigeminovascular axons imbedded in blood vessels of the pia mater and dura mater release CGRP among other vasoactive peptides, producing a sterile inflammatory reaction which in part causes the pain of a migraine attack (Buzzi & Moskowitz, Reference Buzzi and Moskowitz1990; Goadsby et al., Reference Goadsby, Edvinsson and Ekman1990). The mechanisms involved in this inflammatory response have long been the target of migraine studies and data have shown that via T-cell stimulation, cytokines are released when CGRP levels are elevated (Cuesta et al., Reference Cuesta, Quintero, Pons and Suarez-Roca2002; Levite, Reference Levite1998). Furthermore, Levy (Reference Levy2009) showed that the release of inflammatory mediators such as cytokines and mast cells, may further promote and sustain the activation and sensitisation of meningeal nociceptors, inducing the persistent throbbing headache characterized in migraine. Given this link as well as the role of cytokines in modulation of pain and sterile inflammation, these molecules have been proposed as being involved in the pathogenesis of migraine attacks.
Inflammatory Cytokines
Cytokines are signaling molecules that are intricately involved in the activation, differentiation, and control of the immune system. Research has shown that signaling pathways are complex, with many cytokines having multiple functions and acting on different cell types resulting in different functional consequences. Within the broad classification of cytokines, subdivisions are made according to families and subtypes within families (Hiscott & Ware, Reference Ware2011). The tumor necrosis factor (TNF) super family is a class of molecules most involved with activation and inhibition of inflammatory pathways (Ware, Reference Ware2011).
Cytokines are constitutively expressed by neurons and glial cells in brain tissue. Known functions of these regulatory molecules include synaptic plasticity, neural transmission, and Ca2+ signaling. Calcium signaling is thought to have a significant role in migraine as mutations have been identified in the CACNA1A gene that cause the rare migraine subtype FHM (Ophoff et al., Reference Ophoff, Terwindt, Vergouwe, van Eijk, Oefner, Hoffman and Frants1996). It has been shown that these mutations can produce gain-of-function Ca(V) 2.1 channels and as a result initiate CSD. The increased activity of the Ca(V) 2.1 channel facilitates increased Ca(V) 2.1-dependent neurotransmitter release from cortical neurons, in particular glutamate (D'Onofrio et al., Reference D'Onofrio, Ambrosini, Di Mambro, Arisi, Santorelli, Grieco and Buzzi2009).
Given the important regulatory role of inflammatory cytokines, especially in the Ca2+ signaling pathway, it is biologically feasible for dysregulation of the genes involved in these pathways to cause migraine attacks or at least to lower the threshold for triggering one.
Three of the most studied inflammatory cytokines which are thought to play a role in migraine are TNFα, lymphotoxin α (LTA), and lymphotoxin β (LTB). The genes encoding for these products are located in the class III gene cluster of the major histocompatibility complex on chromosome 6 as shown in Figure 1 and variants within these genes have been shown to modulate expression of the corresponding cytokine levels (Bouma et al., Reference Bouma, Crusius, Oudkerk Pool, Kolkman, von Blomberg, Kostense and Pena1996; Carroll et al., Reference Carroll, Katzman, Alicot, Koller, Geraghty, Orr and Spies1987; Wilson et al., Reference Wilson, Symons, McDowell, McDevitt and Duff1997). As seen in Figure 1, the TNF gene cluster is made up of the three genes TNFα, LTA, and LTB. It is thought that these three genes are expressed and regulated in a similar fashion to an operon, hence the reference to them making up a gene cluster.

FIGURE 1 Structure of the LTA gene cluster on chromosome 6.
It is well known that TNFα is an important inflammatory cytokine involved in modulation of immune responses and evidence suggests that it also plays a role in migraine. Biochemical studies have shown that changes in serum and urine concentrations of TNFα correspond to migraine status of patients (Covelli et al., Reference Covelli, Munno, Pellegrino, Di Venere, Jirillo and Buscaino1990; Empl et al., Reference Empl, Sostak, Riedel, Schwarz, Muller, Forderreuther and Straube2003; Mueller et al., Reference Mueller, Gupta and Stein2001; Perini et al., Reference Perini, D'Andrea, Galloni, Pignatelli, Billo, Alba and Toso2005). Furthermore, TNFα has been shown to stimulate transcription of CGRP, which is thought to be critical in the migraine pathogenesis pathway (Durham, Reference Durham2006). Commonly studied polymorphisms are the 308G>A (rs1800629) variant in the promoter region of TNFα and the 252A>G (rs909253) variant in intron 1 of the TNFα gene.
Other variants within the TNF gene cluster region have also been investigated in relation to migraine susceptibility, with conflicting results (Asuni et al., Reference Asuni, Stochino, Cherchi, Manconi, Congiu and Zompo2006; Ghosh et al., Reference Ghosh, Joshi, Pradhan and Mittal2010; Mazaheri et al., Reference Mazaheri, Hajilooi and Rafiei2006; Rainero et al., Reference Rainero, Grimaldi, Salani, Valfre, Rivoiro, Savi and Pinessi2004; Schurks et al., Reference Schurks, Kurth, Buring and Zee2009; Trabace et al., Reference Trabace, Brioli, Lulli, Morellini, Giacovazzo, Cicciarelli and Martelletti2002). Of particular interest was a study conducted in a Korean population, where rs2844482 located in the LTA gene was found to be significantly (p = .0003) associated with migraine (Lee et al., Reference Lee, Jang, Sohn, Won, Kim, Kim and Chung2007). A recent meta-analysis examined the overall association between migraine susceptibility and variants within the TNF gene cluster for a large number of published studies. The authors found no overall association between any of the investigated gene variants and migraine including its subtypes, despite pre-existing biological plausibility. However, most of the studies included in the meta-analysis consisted of small cohorts. The authors suggested that a larger case-control study compared to a single small cohort may provide more statistical power and clarify the potential role of the TNF gene cluster in migraine (Schurks et al., Reference Schurks, Rist, Zee, Chasman and Kurth2011). A recent pedigree Genome-Wide Association Study (pGWAS) conducted in the genetic isolate Norfolk Island population supports this notion, with five variants within the TNF gene cluster found to be nominally significant (Cox, Reference Cox2011; Cox et al., Reference Cox, Lea, Bellis, Carless, Dyer, Curran and Griffiths2012). Three of these variants within the LTA gene namely rs2009658, rs2844482, and rs2229094 that have been subsequently investigated in an outbred Australian Caucasian population (Oikari et al., Reference Oikari, Stuart, Okolicsanyi, Cox, Dixit, Lea and Griffiths2012), however, showed no association with migraine susceptibility.
Given the conflicting nature of the prior studies and the findings by Lee et al. (Reference Lee, Jang, Sohn, Won, Kim, Kim and Chung2007) that haplotypes within the LTA gene demonstrated association with migraine, further investigation of other variants in the LTA gene and additional variants within the TNF gene cluster in a larger case-control cohort was warranted to clarify the involvement of genetic variation in this region. The aim of this study is to expand on previous studies conducted and utilize a larger case-control cohort in addition to investigating a larger range of variants within the TNF gene cluster, specifically in the LTA gene.
Materials and Methods
Sample Selection
Migraine cases and controls were recruited from the local South East Queensland region through the Genomics Research Centre (GRC) outpatient clinic, through various methods including media coverage and advertisements, as well as referral by neurologists (Colson et al., Reference Colson, Lea, Quinlan, MacMillan and Griffiths2004). They were all of Caucasian origin, and diagnosed as having MA or MO based on criteria specified by the International Headache Society. Each participant was required to fill out standardized questionnaires outlining frequency and severity of migraine attacks as well as family history. This information was used to make a diagnosis. An unaffected control group with no family history of migraine was matched for age (+/- 5 years), sex, and ethnicity. Blood samples obtained from patients were collected through the GRC clinic and DNA was extracted using a salting out method. Approval for the study protocol was acquired from the Griffith University's Ethics Committee for Experimentation on Humans. A total of 736 samples were genotyped for nine variants found within the LTA and TNFα genes as follows: rs1800683, rs2229094, rs2009658, rs2071590, rs2239704, rs909253, rs1800630, rs1800629, and rs3093664. Of these 736 samples, 192 samples were genotyped for the single nucleotide polymorphisms (SNPs) rs2229094 and rs2009658 in a previous study (Oikari et al., Reference Oikari, Stuart, Okolicsanyi, Cox, Dixit, Lea and Griffiths2012). The remaining 544 samples were genotyped for the first time for these variants. The other seven SNPs selected were not genotyped previously in any of the 736 samples. One SNP previously genotyped, namely rs2844482, was not included due to incompatibility in the SNP plex designed for Sequenom.
Molecular Analysis
Matrix-assisted desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was used to genotype each sample in a multiplexed reaction (Shchepinov et al., Reference Shchepinov, Denissenko, Smylie, Worl, Leppin, Cantor and Rodi2001). The Sequenom instrument and accompanying software were used to carry out all genotyping work as well as the design of amplification and genotyping primers. In this approach, unique primer pairs were designed to amplify each region of interest such that only one region would be amplified for each primer pair. A genotyping primer was then designed to anneal directly adjacent to the SNP of interest, allowing for enzymatic extension of a single dideoxy base pair following polymerase chain reaction amplification. The extended primer was then robotically dispensed onto a silicon chip preloaded with matrix, ionized and released. The time-of-flight was recorded for each fragment, which has a slightly different density according to genotype and interpreted as a scatter plot. Genotyping was confirmed independently using either high-resolution melt (HRM) analysis (rs2229094) or restriction fragment length polymorphism (RFLP) analysis (rs2009658; Oikari et al., Reference Oikari, Stuart, Okolicsanyi, Cox, Dixit, Lea and Griffiths2012).
Statistical Analysis
Of the 736 samples genotyped, 25 samples failed due to poor quality (3%) and 26 samples were included as duplicate or triplicate controls of other samples. Genotyping of these samples was removed prior to analysis after confirmation that genotyping results matched across samples. After filtering, 685 genotyped individuals remained, which included 167 males and 518 females divided into 340 unique cases and 345 controls.
SPSS was used to perform a basic allelic association analysis using a χ2 test, comparing the allele frequencies of migraine cases to controls for each SNP investigated. Hardy–Weinberg equilibrium (HWE) was calculated for each SNP to check for genotyping error and an α value of 0.005 was set as the significance threshold to correct for multiple tests. Migraine cases were further subdivided and compared to controls according to the subtypes MA and MO. Analysis according to sex was also performed. The odds ratio was calculated with a 95% confidence interval as an indication of disease risk. The minor allele frequency was calculated and compared to European HapMap frequencies. Haploview was used to determine linkage disequilibrium (LD) blocks across the two genes investigated.
Results
HWE calculations showed that each SNP was in HWE in both cases and controls, except for one SNP, rs1800629, which gave a P value of .04 in the case samples. Given that the deviation from HWE was so small and also that the accuracy of genotyping results was confirmed through independent HRM and RFLP analysis, these data were still included in our analysis.
Allele frequencies for both cases and controls were compared to HapMap European population data, and there are no significant differences between frequency values. Genotyping results showed that the variants tested for in our Australian Caucasian population occur at similar frequencies compared to other European populations.
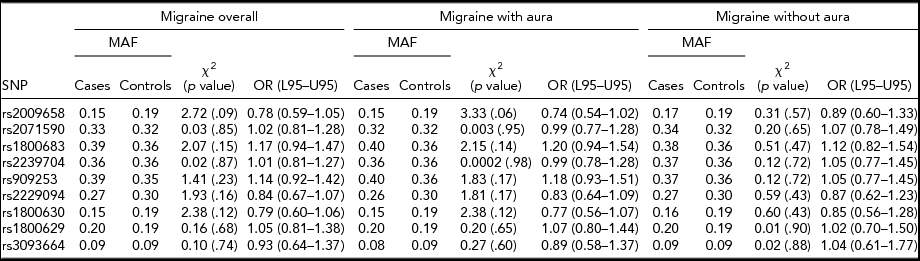
An allelic association test showed that the allele frequencies were not significantly different between overall migraine cases and controls for all nine SNPs tested. When migraine was analyzed according to subtype, no significant difference was found between MA compared to controls and MO compared to controls for any of the SNPs tested, as illustrated in Table 1. No significant association was found between migraine susceptibility and sex. The odds ratios calculated for each test did not convey a significant risk or protective effect for migraine susceptibility for any of the SNPs tested. Gender and migraine subtype also was not found to influence disease risk for the nine SNPs tested.
TABLE 1 Association Between LTα and TNFα Variants and Migraine Susceptibility According to Migraine Subtypes, and Migraine Overall
Haplotype analysis using Haploview showed that of the nine variants tested, eight were in almost complete LD, as illustrated in Figure 2. Markers were grouped into two haplotype blocks, with block one containing eight of the nine markers. Association testing according to haplotype revealed no significant association between migraine and any of the haplotypes as shown in Table 2.
TABLE 2 Haplotype Association Testing Using Haploview

FIGURE 2 LTA SNPs tested for linkage disequilibrium with results given by D' using Haploview.
Discussion
Both IL-1 and TNFα are well-studied cytokines in the field of molecular medicine (Feuerstein et al., Reference Feuerstein, Liu and Barone1994; Hiscott & Ware, Reference Ware2011). Particularly, popular choices in migraine susceptibility studies include these molecules in addition to variants found within the TNF gene cluster. Previous studies have shown that both TNFα and LTA could be susceptibility genes in migraine, and it has been suggested that other functional polymorphisms that influence migraine risk could be in LD with some of these variants. However, studies have produced conflicting results and have also shown that there are significant differences in allele frequencies of TNFα gene variants between different ethnic groups (Lee et al., Reference Lee, Jang, Sohn, Won, Kim, Kim and Chung2007; Schurks et al., Reference Schurks, Rist, Zee, Chasman and Kurth2011). A previous study found that while individual SNPs within the TNF gene cluster were not significantly associated with migraine, LTA haplotypes were associated with migraine in the Korean population. Authors also suggested that associations would be best defined in context with the involvement of other genetically linked regions, such as HLA class I loci (Lee et al., Reference Lee, Jang, Sohn, Won, Kim, Kim and Chung2007).
The aim of this study was to expand on previous studies conducted and investigate previously associated variants as well as other suitable markers within the TNF gene cluster in relation to migraine susceptibility in an Australian Caucasian population. Our selection of nine SNPs across this region allowed for more detailed haplotype analysis, in addition to case-control association analysis. While HWE calculations confirmed genotyping accuracy, no significant association was found with migraine in any of the statistical tests conducted. Our results do, however, illustrate the value of testing multiple markers as multiple SNPs provide more information than single SNPs for the purposes of complex disease mapping. Future studies which examine multiple cytokine family gene variants in conjunction with linked HLA class I haplotypes would be useful.
No significant association was found between the tested variants and migraine susceptibility in this case-control Australian Caucasian population, even when analysis was done in accordance with migraine subtypes and according to haplotypes. This result suggests that either the TNF gene cluster does not play a role in the pathogenesis of migraine, or that it only plays a minor role as part of a more complex signaling cascade. Given the large diversity of cytokines, it would be useful to examine other families within this broad class of signaling molecules and the role that they may play in sterile inflammation during a migraine attack. A multi-marker analysis approach, making use of other linked HLA regions, would also provide better insight into the role of cytokines during the inflammation process. A meta-analysis that incorporates all the studies conducted to date would also be a useful tool to conclude whether any of the TNF markers are associated with migraine.
A number of large GWAS aiming to identify associations between genetic variants and migraine have been conducted, with the first being published by the International Headache Genetics Consortium (IHGC) in 2010 (Anttila et al., Reference Anttila, Stefansson, Kallela, Todt, Terwindt, Calafato and Palotie2010; Schurks, Reference Schurks2012). Two subsequent GWAS conducted by the Dutch-Icelandic migraine genetics consortium (DICE) and the Women's Genome Health Study (WGHS) failed to replicate the results from the first GWAS and only identified modest associations (Ligthart et al., Reference Ligthart, de Vries, Smith, Ikram, Amin, Hottenga and Boomsma2011). However, a large meta-analysis of all the cohorts identified three new SNPs showing significance at the genome-wide level, namely rs2651899, rs10166942, and rs11172113 located in intron 1 of PRDM16, 2q37 near TRPM8 and intron 1 of LRP1, respectively (Chasman et al., Reference Chasman, Schurks, Anttila, de Vries, Schminke, Launer and Kurth2011).
Thus far, SNPs in the TNF gene cluster have not shown genome-wide significance in these large GWAS. However, given the complex inheritance and the gene–environment interactions known to occur for complex diseases, a very large sample size would be required before variants with small effects are detected. More targeted family linkage studies and association studies could provide a useful approach for detecting variants with small to moderate effects on migraine susceptibility. Gene expression studies have shown significant changes in expression of TNF for menstrual migraineurs, so while association could be limited to specific subtypes of migraine, future studies are still relevant (Mueller et al., Reference Mueller, Gupta and Stein2001).
Acknowledgments
This work was funded by an NHMRC grant. Contributing authors were supported by Australian Post-graduate Award (APA) scholarships and Griffith University Postgraduate Award (GUPA) scholarships.