1. An introduction to the kink-turn (k-turn) in RNA structure
The k-turn is a common structural motif in many functional RNA species, that introduces a sharp bend into the axis of duplex RNA. In most cases, k-turn structures act as binding sites for specific proteins and frequently mediate short- or long-range tertiary contacts. The secondary structures of medium to large RNA species can be reduced to double-helical sections connected by junction elements that alter the axial trajectory and allow tertiary interactions to occur. The k-turn is probably the most common such element in RNA. The structural and folding characteristics of k-turns and their dependence on local sequence have been studied extensively, such that they are arguably the best understood of the motifs from which RNA is built.
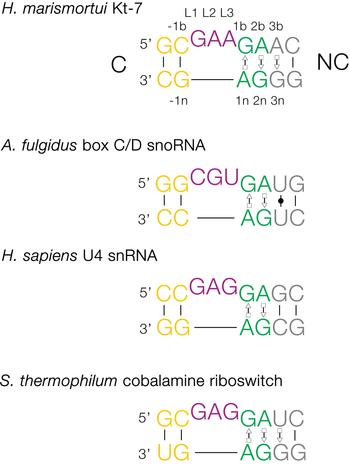
The basic, standard k-turn comprises a (usually) three-nucleotide bulge followed on the 3′ side by tandem G:A and A:G pairs (Fig. 1). The helices 5′ and 3′ to the loop are termed the C- and NC-helices, respectively (names derived from the presence of canonical or non-canonical base pairing). The standard nomenclature (Liu & Lilley, Reference Liu and Lilley2007) for the nucleotide positions numbers outwards from the ends of the loop, positively in the 3′ direction and negatively in the 5′ direction. The nucleotides located on the loop-containing strand (that is normally drawn as the top strand) take the suffix b, and those on the other strand take the suffix n. The nucleotides of the loop are named L1, L2 and so on numbered from the 5′ end. In a subset of k-turns, termed k-loops, the C helix is replaced by a terminal loop with no formal base-pairing.

Fig. 1. Sequences of some representative simple k-turns and the nomenclature of the nucleotide positions. Four examples of k-turns are shown, drawn from the large ribosomal subunit (Kt-7), box C/D snoRNP, the spliceosomal B complex U4 snRNA and a (cobalamine) riboswitch. The nomenclature is shown for Kt-7. The helix 5′ to the loop is called C, and that to the 3′ is called NC. The loop nucleotides are designated Ln numbered from 5′ to 3′. For the non-loop nucleotides, those on the loop strand take the suffix b and those on the other strand take n. The nucleotides of the NC helix are positively numbered outwards (i.e. 5′ to 3′ on the loop-containing strand) from the loop, while those of the C helix are negatively numbered also outward from the loop (3′ to 5′ on the loop-containing strand). When this nomenclature is applied to complex k-turns the nucleotides are numbered according to their positions in the structure rather than their order in the primary sequence. The coloring of the nucleotides is followed throughout this review.
k-turns kink the helical axis by juxtaposing the minor grooves of the NC and C helices, with the major groove displayed on the outer face of the structure. The helical axes are not exactly coplanar, but there is an included angle of 50° in projection. This angle is quite tightly constrained by the hydrogen bonds that form in the core, and thus the angle between the axes for the known k-turns occupies a narrow range between 45° and 55° (Daldrop & Lilley, Reference Daldrop and Lilley2013).
The structure is stabilized by important A-minor H-bonding interactions between the conserved adenine nucleobases of the tandem G•A pairs and 2-hydroxyl groups of the C-helix and loop. These are discussed further in section 3.
2. The occurrence of k-turns
k-turns are extremely widespread, being found in most classes of functional RNA in all domains of life. They were identified as a common sequence element in RNA (Winkler et al. Reference Winkler, Grundy, Murphy and Henkin2001), but first recognized as a recurring structural motif when Steitz and coworkers solved the crystal structure of the large ribosomal subunit of the archaeon Haloarcular marismortui (Ban et al. Reference Ban, Nissen, Hansen, Moore and Steitz2000; Klein et al. Reference Klein, Schmeing, Moore and Steitz2001). k-turns are also found in the small ribosomal subunit (Wimberly et al. Reference Wimberly, Brodersen, Clemons, Morgan-Warren, Carter, Vonrhein, Hartsch and Ramakrishnan2000), and in the ribosomes of bacteria (Clemons et al. Reference Clemons, May, Wimberly, Mccutcheon, Capel and Ramakrishnan1999; Schuwirth et al. Reference Schuwirth, Borovinskaya, Hau, Zhang, Vila-Sanjurjo, Holton and Cate2005) and eukaryotes (Ben-Shem et al. Reference Ben-Shem, Garreau, Loubresse, Melnikov, Jenner, Yusupova and Yusupov2011). Most ribosomal k-turns mediate long- or short-range tertiary contacts and the majority are bound by proteins. Kt-7 of the H. marismortui 50S ribosomal subunit (Klein et al. Reference Klein, Schmeing, Moore and Steitz2001) has a sequence that is close to the consensus for standard k-turns (Fig. 1), and is the best characterized of any k-turn despite the fact that it is atypical in one respect (see section 4.1); we shall refer to this as HmKt-7 subsequently.
A k-turn plays a role in the assembly of the eukaryotic spliceosome, found in U4 snRNA that is contained within the B-complex that precedes the tri-snRNP complex (Vidovic et al. Reference Vidovic, Nottrott, Hartmuth, Luhrmann and Ficner2000; Wozniak et al. Reference Wozniak, Nottrott, Kuhn-Holsken, Schroder, Grubmuller, Luhrmann, Seidel and Oesterhelt2005). The U4 k-turn binds the 15·5k protein, the eukaryotic ortholog of the L7Ae protein (see section 8). In an ordered assembly process (Nottrott et al. Reference Nottrott, Urlaub and Luhrmann2002) the complex then recruits Prp31 (Liu et al. Reference Liu, Li, Dybkov, Nottrott, Hartmuth, Luhrmann, Carlomagno and Wahl2007) in a manner closely analogous to the assembly of the box C/D snoRNP discussed below (section 12). This complex can be observed in the recent cryo-EM structure of the spliceosomal B complex of yeast (Plaschka et al. Reference Plaschka, Lin and Nagai2017). Given the size and complexity of the spliceosome, it seems quite possible there will be further k-turns formed at different stages of the spliceosome cycle.
k-turns are important structural elements in at least seven different riboswitches. The first was discovered in the SAM-I riboswitch (Montange & Batey, Reference Montange and Batey2006), and examples were later found in the structures of the cyclic-diGMP (Smith et al. Reference Smith, Lipchock, Ames, Wang, Breaker and Strobel2009, Reference Smith, Shanahan, Moore, Simon and Strobel2011), cobalamine (Peselis & Serganov, Reference Peselis and Serganov2012) and T-box (Zhang & Ferre-D'Amare, Reference Zhang and Ferre-D'amare2013) riboswitches. Biochemical experiments have also strongly indicated the presence of k-turns in lysine (Blouin & Lafontaine, Reference Blouin and Lafontaine2007) and glycine (Baird & Ferre-D'Amare, Reference Baird and Ferre-D'amare2013) riboswitches. Lastly, bacterial and plant thiamine pyrophosphate (TPP) riboswitches have an elaborated form of k-turn called a k-junction (Serganov et al. Reference Serganov, Polonskaia, Phan, Breaker and Patel2006; Thore et al. Reference Thore, Leibundgut and Ban2006; Wang et al. Reference Wang, Daldrop, Huang and Lilley2014), discussed below in section 6.3. The riboswitches are not generally known to bind proteins, and their role appears to be the mediation of tertiary contacts to create a stable fold (and thereby a ligand binding site) in these relatively small, autonomously folding RNA species.
k-turns play a critical role in the assembly of the box C/D and H/ACA snoRNP structures that carry out guided O2′-methylation and pseudouridylation, respectively, of archaeal and eukaryotic RNA (Hamma & Ferré-D'Amaré, Reference Hamma and Ferré-D'amaré2004; Moore et al. Reference Moore, Zhang, Fenley and Li2004; Szewczak et al. Reference Szewczak, Gabrielsen, Degregorio, Strobel and Steitz2005; Youssef et al. Reference Youssef, Terns and Terns2007). k-turn-forming sequences are also found in U3 snoRNP involved in guided nucleolytic processing of rRNA (Beltrame & Tollervey, Reference Beltrame and Tollervey1995; Gerbi et al. Reference Gerbi, Borovjagin, Ezrokhi and Lange2001; Marmier-Gourrier et al. Reference Marmier-Gourrier, Clery, Senty-Segault, Charpentier, Schlotter, Leclerc, Fournier and Branlant2003). The core of the box C/D snoRNA forms an internal loop, the strands of which provide the 12 nt guide sequences for two target RNA species. The loop is flanked by the box C and D, and box C′ and D′ sequences that form k-turns or k-loop structures at each end (Bleichert et al. Reference Bleichert, Gagnon, Brown, Maxwell, Leschziner, Unger and Baserga2009; Kiss-Laszlo et al. Reference Kiss-Laszlo, Henry, Bachellerie, Caizergues-Ferrer and Kiss1996; Lin et al. Reference Lin, Lai, Jia, Xu, Zhang, Lu and Ye2011; Tran et al. Reference Tran, Zhang and Maxwell2003; Tycowski et al. Reference Tycowski, Smith, Shu and Steitz1996; Watkins et al. Reference Watkins, Segault, Charpentier, Nottrott, Fabrizio, Bachi, Wilm, Rosbash, Branlant and Luhrmann2000; Xue et al. Reference Xue, Wang, Yang, Terns, Terns, Zhang, Maxwell and Li2010; Ye et al. Reference Ye, Jia, Lin, Ju, Peng, Xu and Zhang2009). The k-turns bind L7Ae-family protein (L7Ae in archaea, 15·5k in eukaryotes) as the first stage of snoRNP assembly (McKeegan et al. Reference Mckeegan, Debieux, Boulon, Bertrand and Watkins2007; Omer et al. Reference Omer, Ziesche, Ebhardt and Dennis2002; Schultz et al. Reference Schultz, Nottrott, Watkins and Luhrmann2006; Watkins et al. Reference Watkins, Newman, Kuhn and Maxwell1998, Reference Watkins, Dickmanns and Luhrmann2002). This will be discussed further in section 12.
k-turns play a significant role in the control of translation of some mRNA species. A k-turn was found in a stem-loop contained within the pre-mRNA encoding the L30e ribosomal protein of Saccharomyces cerevisiae (Mao et al. Reference Mao, White and Williamson1999; White et al. Reference White, Hoeger, Schweppe, Shillingford, Shipilov and Zarutskie2004). L30e is a member of the L7Ae family of proteins, and binds a k-turn within its pre-mRNA, thereby repressing its own translation. It has recently been shown that in archaea L7Ae similarly regulates its own translation by binding to a k-turn within the 5′-UTR of its structural gene l7ae, thereby occluding the Shine-Dalgarno sequence (Daume et al. Reference Daume, Uhl, Backofen and Randau2017). This may be a more general mechanism, as L7Ae has been shown to bind to mRNA encoding the box C/D snoRNP components Nop5 and fibrillarin (Daume et al. Reference Daume, Uhl, Backofen and Randau2017) (for discussion of box C/D assembly see section 12).
Thus k-turns are key architectural elements involved in some of the most important functions of RNA in the cell. These include translation, various kinds of posttranscriptional modification including splicing, 2′-O-methylation and pseudouridylation, and in the control of gene expression. And the list does not stop there. For example, a k-turn occurs in the ribozyme ribonuclease P that processes the 5′-terminus of tRNA (Lai et al. Reference Lai, Tanimoto, Lai, Chen, Marathe, Westhof, Wysocki and Gopalan2017; Reiter et al. Reference Reiter, Osterman, Torres-Larios, Swinger, Pan and Mondragon2010). Still more k-turns exist of currently unknown function. For example, Breaker and colleagues (Weinberg et al. Reference Weinberg, Lunse, Corbino, Ames, Nelson, Roth, Perkins, Sherlock and Breaker2017) have recently presented a bioinformatic analysis of 224 candidate structured RNAs found in non-coding regions of bacteria, a number of which contain putative k-turn structures – see section 6.5. Thus it is highly probable that many more are waiting to be discovered, for example in the analysis of long non-coding RNA species.
3. The structure of a standard k-turn
Tandem G:A, A:G basepairs are the core of the k-turn structure, each forming trans G(sugar edge):A(Hoogsteen edge) base pairs (Fig. 2). The 1b:1n pair forms H-bonds from GN2 to AN7, and from AN6 to GN3. The 2b:2n pair is similar, except that in a subset of k-turns called the N1 class (see section 4 below) the AN6 to GN3 is too long to be considered a stable H-bond. The nucleobase of L1 is stacked onto the end of the C-helix, while that of L2 is stacked onto the end of the NC helix. The 1b:1n pair is strongly buckled such that G1b is rotated ~25° out of the plane, and L2 adopts a syn conformation about the glycosyl bond in order to maximize stacking on A1n. L3 is directed away from the RNA structure into the solvent, making no interactions within the RNA.

Fig. 2. The structure of a simple k-turn. (a) The sequence of HmKt-7 with the key cross-strand H-bonds indicated by the broken cyan arrows. These are donated by the 2′-hydroxyl groups of the L1 and −1n ribose groups, and accepted by the conserved adenine nucleobases at the 1n and 2b positions, respectively. (b) and (c) Parallel-eye stereoscopic image of the structure of HmKt-7. The k-turn structure was determined as a duplex at 2·0 Å resolution and is in the N3 conformation (protein data base (PDB) ID 4CS1). The overall structure of the k-turn viewed from the side of the loop is shown in (b), while the G:A basepairs and the cross-strand H-bonds are shown in (c). All H-bonds are shown as broken lines here and throughout the review. (d) The chemical structure of the sheared trans-sugar-Hoogsteen G:A basepairs.
In the folded k-turn structure the minor grooves of the NC and C helices are juxtaposed, with the sugar edges of the conserved adenine nucleobases (1n and 2b) directed towards the minor groove of the C helix. These act as acceptors for critical cross-strand H-bonds donated by 2′-OH groups (Fig. 2). A1n N1 accepts a proton from the O2′ of L1 (the 5′ nucleotide of the loop) (Lescoute et al. Reference Lescoute, Leontis, Massire and Westhof2005; Liu & Lilley, Reference Liu and Lilley2007), while A2b N1 or N3 (see the following section) accepts one from the O2′ of the nucleotide at −1n (Daldrop & Lilley, Reference Daldrop and Lilley2013; Reblova et al. Reference Reblova, Sponer, Spackova, Besseova and Sponer2011). These two H-bonds are extremely important to the integrity of the folded structure. Disruption of the L1 to A1n H-bond was shown to prevent the folding of HmKt-7 in response to the addition of Mg2+ ions (Liu & Lilley, Reference Liu and Lilley2007). In k-turn variants that disallow one or other then surrogate equivalents form. For example, HmKt-58 has a loop of just two nucleotides and thus it is the O2′ of the nucleotide in the −1b position that donates a proton to A1n N1 (Liu & Lilley, Reference Liu and Lilley2007). Additional H-bonds form adventitiously in given k-turns. In many k-turns L3 O2′ donates an H-bond to the pro-S non-bridging O of the L1/L2 phosphate; removal of the L3 2′-OH from HmKt-7 led to impaired ion-induced folding (Liu & Lilley, Reference Liu and Lilley2007).
4. Two conformational classes adopted by k-turns
Comparison of the available structures of k-turns reveals that the H-bond donated by −1n O2′ can be accepted by A2b at either N3 or N1 (Fig. 3). This divides the known k-turns into two classes termed N3 or N1, with approximately equal numbers in each group (Table 1). In order to accept a proton at N1 (i.e. adopt the N1 conformation), the nucleobase of A2b must rotate, stretching the A2b N6 to G2n N3 distance typically to >4 Å, too long to be a stable H-bond. This is compensated in the great majority of N1-class k-turns by the formation of a second H-bond from by G-1n N2 to A2b N3, with good length and geometry. The switch between the N3 and N1 conformations leads to a rotation about the C helix axis (Daldrop & Lilley, Reference Daldrop and Lilley2013), potentially affecting any tertiary interaction that the helix makes.

Fig. 3. The N3 and N1 conformations of the k-turn. (a) A parallel-eye stereoscopic image of the structure of the core of the ribosomal k-turn HmKt-38 core as a representative N1 structure (PDB ID 1FFK). Note that the G1b:A1n basepair has the standard two H- bonds, while the A2b:G2n nucleobases are only connected by a single H-bond from G2nN2 to A2bN7. The distance between A2bN6 and G2nN3 is 4·7 Å (shown by the broken red line) is too long to be stably H-bonded. (b) and (c) A comparison of the H-bonding in the 2n:2b:-1n triple–nucleotide interaction in HmKt-7 in the N1 (b, in the ribosome; PDB ID 1FFK) and N3 (c, as a duplex PDB ID 4CS1). conformations. Note again the long A2bN6 to G2nN3 distance in the N1 conformation, shown as a broken red line in b.
Table 1. Summary of the sequence and conformation of simple, standard k-turns

The name of each k-turn (and binding protein where relevant) are shown, above the organism of origin.
The PDB ID and resolution for each structure are shown, and the space group in which the structure was solved. The −1b:−1n, loop and 3b:3n sequences are tabulated, together with the conformation of the 3b:3n as either cis-Watson-Crick (cWC) or trans-sugar-Hoogsteen (tSH). Lastly, the conformation as N3 or N1 type is shown for each k-turn. rsw, riboswitch.
4.1 HmKt-7 can exist in both N3 and N1 conformations
The structural class for a given k-turn is generally invariant, irrespective of its environment. However, there is an interesting exception to this. In the H. marismortui large ribosomal subunit Kt-7 adopts the N1 conformation (Ban et al. Reference Ban, Nissen, Hansen, Moore and Steitz2000). Yet when taken out of the ribosomal context it invariably adopts the N3 conformation, as free RNA (Daldrop & Lilley, Reference Daldrop and Lilley2013; Huang & Lilley, Reference Huang and Lilley2013) or bound by L7Ae protein (Huang & Lilley, Reference Huang and Lilley2013). In fact aside from the ribosome, 16 independent structures (some being crystallographically-independent molecules within the same lattice) have been determined for HmKt-7, all of which are N3 structures (Huang et al. Reference Huang, Wang and Lilley2016). Thus it seems that the environment of the archaeal ribosome has forced Kt-7 to adopt the N1 conformation even though this is clearly less stable in isolation compared with the N3 structure. In the ribosome, Kt-7 is contained within a duplex that is organized by a three-way junction and whose terminal loop makes a loop–loop interaction with an adjacent arm of the junction. It is also bound by the L24 protein. Evidently, these interactions increase the relative stability of the N1 conformation in the ribosomal context. There is one other example of conformational switching known. The conformation of the non-standard (see section 6.1) ribosomal k-turn Kt-23 from Thelohania solenopsae was determined to be N1 class in the context of the SAM-I riboswitch, but N3 when bound to L7Ae (Huang & Lilley, Reference Huang and Lilley2014).
5. The conformational class is determined by the 3b,3n sequence
The structural class of most k-turns is intrinsic, unaffected by context. A given k-turn will adopt the same conformation as free RNA or protein bound (Huang & Lilley, Reference Huang and Lilley2013), when contained within the ribosome, the SAM-I riboswitch or bound by L7Ae (Huang et al. Reference Huang, Wang and Lilley2016). Thus k-turn conformation is evidently determined by the nucleotide sequence. Given the strong conservation of most nucleotides of the standard k-turn (the tandem G:A, A:G pairs and very frequently −1b:−1n = C:G) the possible location of the determinant of conformation is very limited. We found that the key determinant was the 3b:3n position. The known k-turn structures can be plotted in a 4 × 4 array of the 16 possible 3b:3n sequences (Fig. 4) (Huang et al. Reference Huang, Wang and Lilley2016), whereupon it is apparent that a given 3b:3n sequence is associated with either N3 or N1 conformation, except for HmKt-7 as discussed above. Moreover, this holds true irrespective of the sequence context of the k-turn. For example, three different k-turns are known with 3b:3n = U:G, one contained within the cobalamine riboswitch (Peselis & Serganov, Reference Peselis and Serganov2012), one within Thermus thermophilus ribosome and one bound by L7Ae protein, and all three adopt the N1 conformation. Three k-turns have 3b:3n = G:C, and all three adopt the N3 conformation. Similarly there are three k-turns with 3b:3n = U:U, all of which are in the N3 conformation. All the natural k-turns occupy eight cells of the 4 × 4 array, so a systematic synthetic approach was adopted to determine the preferred conformations of the remaining 3b:3n sequences where possible. HmKt-7 inserted in place of the natural SAM-I riboswitch k-turn was systematically modified at the 3b:3n position. Each was placed in crystallization trials resulting in structures determined at 1·71–3·31 Å resolution for 12 of the possible 16 sequences (Huang et al. Reference Huang, Wang and Lilley2016). These additional structures are included (red) in Fig. 4. Of these six fill previously empty cells of the array, leaving only two unoccupied cells. The six remaining structures fall into cells with pre-existing data from the natural k-turns; in every case the newly-determined structures adopt the same conformation as the other members of the same cell. This study also demonstrates that the SAM-I riboswitch is good vehicle for studying k-turn structure, being flexible enough not to perturb the natural conformation of a given k-turn.
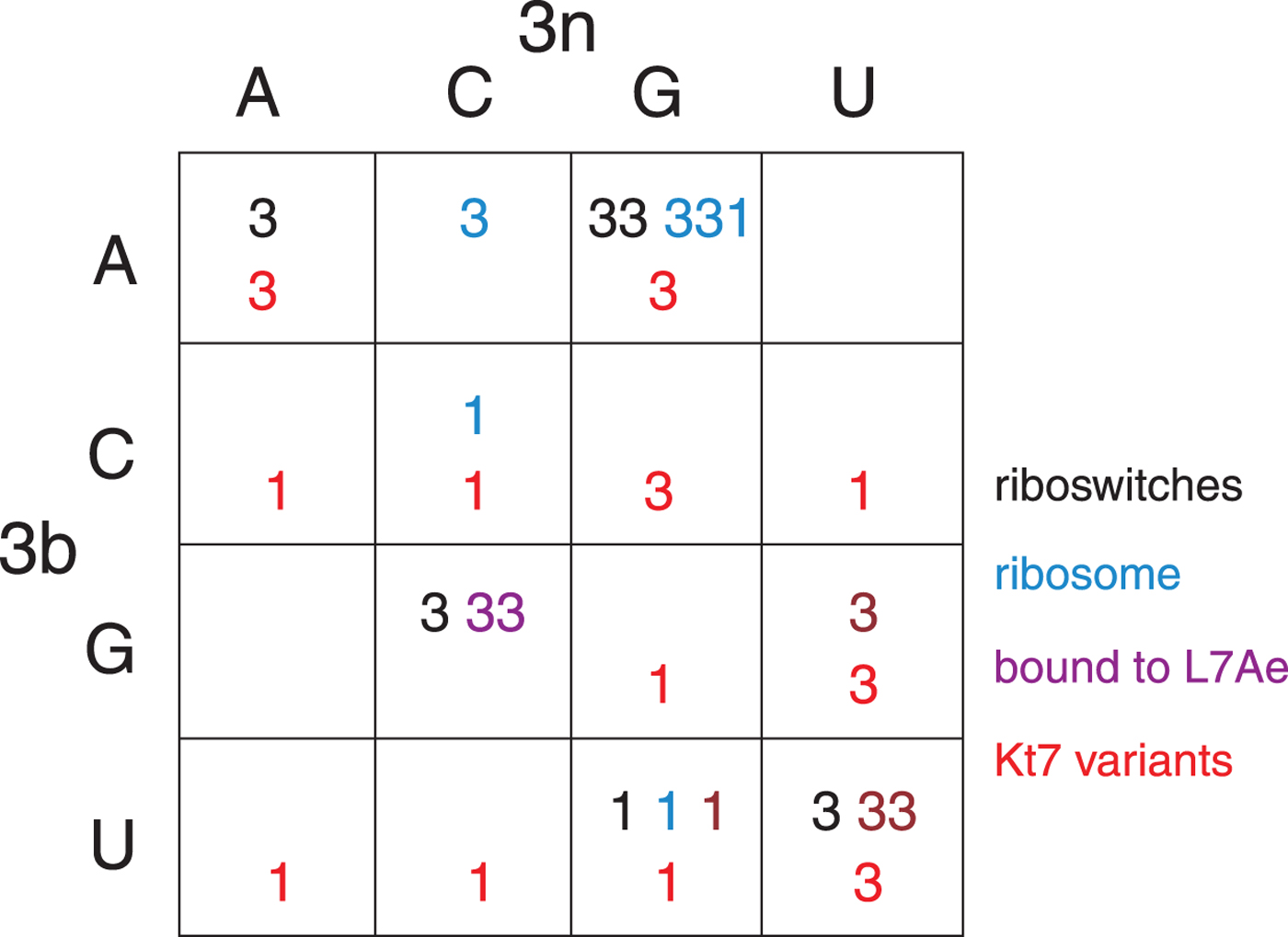
Fig. 4. The conformation of k-turns as a function of the 3b:3n sequence, shown as a 4 × 4 array where the rows show the 3b and the columns the 3n nucleotides (Huang et al. Reference Huang, Wang and Lilley2016). The Watson-Crick base pairs are found on the ascending diagonal. The N3 (3) or N1 (1) conformation is shown for each k-turn. These structures have been determined for riboswitches (black), within the ribosome (cyan), as complexes with L7Ae protein (magenta) and as 3b:3n sequence variants of HmKt-7 (red). Note that where multiple k-turns have the same 3b:3n sequence these, in general, adopt the same conformation. The one exception to this is found in the 3b:3n = A:G cell, where a single case has adopted the N1 conformation. This is HmKt-7 in the context of the ribosome where the environment evidently forces the k-turn to adopt a less favorable conformation. In all other circumstances, HmKt-7 adopts the N3 conformation.
In the HmKt-7 context five of the 3b:3n variants retain the N3 conformation, while seven switch to the N1 conformation. We conclude that the 3b:3n position is the major sequence determinant of k-turn conformation. The full array (Fig. 4) can be used to predict the conformation of new k-turns. However, there is no clear pattern that reveals itself, nor a set of rules relating 3b:3n sequence to conformation. The 3b:3n nucleotides adopt two kinds of basepair, either cis-Watson-Crick or trans-Hoogsteen-sugar edge pairs. Yet there is no apparent correlation of the 3b:3n basepair type with k-turn conformation. However, there is a structural correlation based on base pair width. The rotation of the A2b nucleobase required to accept a donated proton at N1 changes the width of the base pair. Taking the ensemble of k-turn structures available, the mean C1′-C1′ distance for the 2b:2n base pair for the N3 conformation structures is 8·93 ± 0·22 Å. However, that for the N1 conformation is 10·17 ± 0·40 Å. Thus the local helical width at the 2b:2n base pair is significantly greater for the N1 conformation k-turns. If we plot the C1′-C1′ distances for the 2b:2n base pairs against those for the 3b:3n base pairs for the entire set of k-turn structures we find that they cluster into two distinct groups (Fig. 5); the sets of N3 and N1 structures are very nearly disjoint, 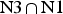 ${\rm N3}\mathop \cap \nolimits {\rm N1}$ is almost empty. The N3 structures have shorter values of both C1′-C1′ distances, while the N1 structures have longer values for both. Interestingly, there is one N1 structure that lies with the N3 structures on this plot, that of HmKt-7 in the ribosome. This reveals its true character because it is intrinsically an N3 k-turn that has been forced to adopt the N1 structure in the ribosome (see section 4). Ribosomal HmKt-7 is very much the exception that proves the rule.
${\rm N3}\mathop \cap \nolimits {\rm N1}$ is almost empty. The N3 structures have shorter values of both C1′-C1′ distances, while the N1 structures have longer values for both. Interestingly, there is one N1 structure that lies with the N3 structures on this plot, that of HmKt-7 in the ribosome. This reveals its true character because it is intrinsically an N3 k-turn that has been forced to adopt the N1 structure in the ribosome (see section 4). Ribosomal HmKt-7 is very much the exception that proves the rule.
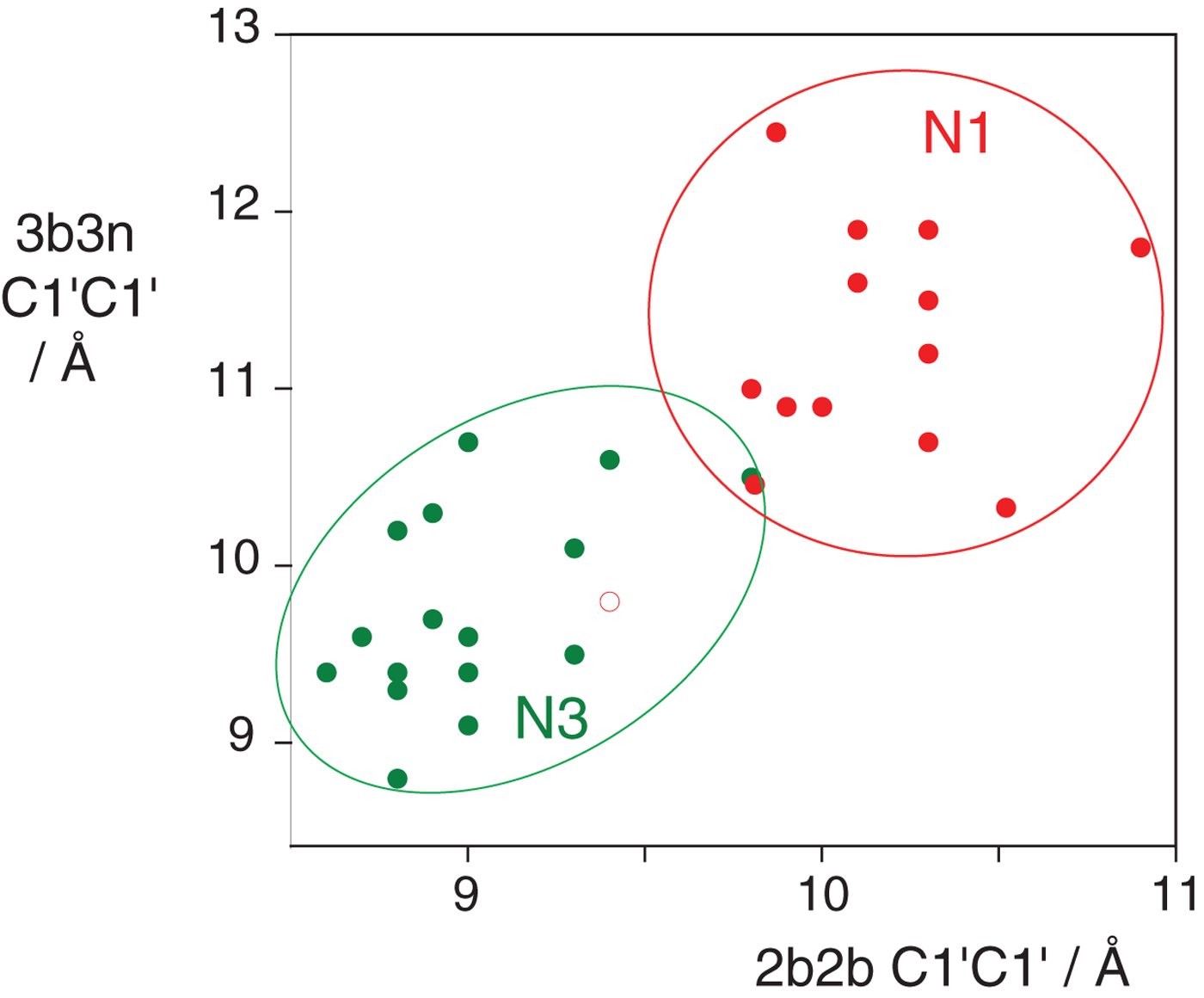
Fig. 5. A plot of C1′-C1′ distances for the 2b:2n and 3b:3n basepairs (Huang et al. Reference Huang, Wang and Lilley2016). These are colored red for N1 structures and green for N3 structures. Note that these form two distinct clusters with longer distances for both basepairs for the N1 structures compared with the N3 structures. Note however that the ribosomal HmKt-7 k-turn (shown as a red open circle) falls within the N3 cluster.
6. Variations on the structural theme of the standard k-turn
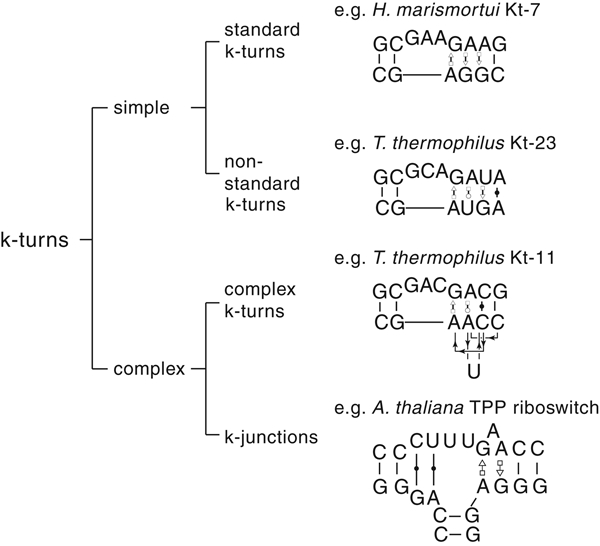
The most common form of the k-turn motif has a three-nucleotide bulge followed by the tandem G:A, A:G pairs, but there is some variation found in natural sequences. We have already noted that in the ribosome some k-turns depart from the standard k-turn motif, with loops that differ from the usual 3 nt, and some have a nucleotide on the lower strand opposing the bulge. But even the G:A, A:G pairs are not completely invariant. Our classification scheme is shown in Fig. 6. Simple k-turns retain the basic framework of the k-turn, whereas in the complex k-turns the nucleotides of the G:A, A:G pairs do not map linearly onto the RNA sequence. Within these groups, non-standard k-turns have a nucleotide substitution within the G:A, A:G pairs. In k-junctions, the k-turn has been elaborated into a helical junction by the presence of an additional helix. While these variations from the standard k-turn reflect significant changes in sequence, we find that in general all the standard structural features of the k-turn are retained.
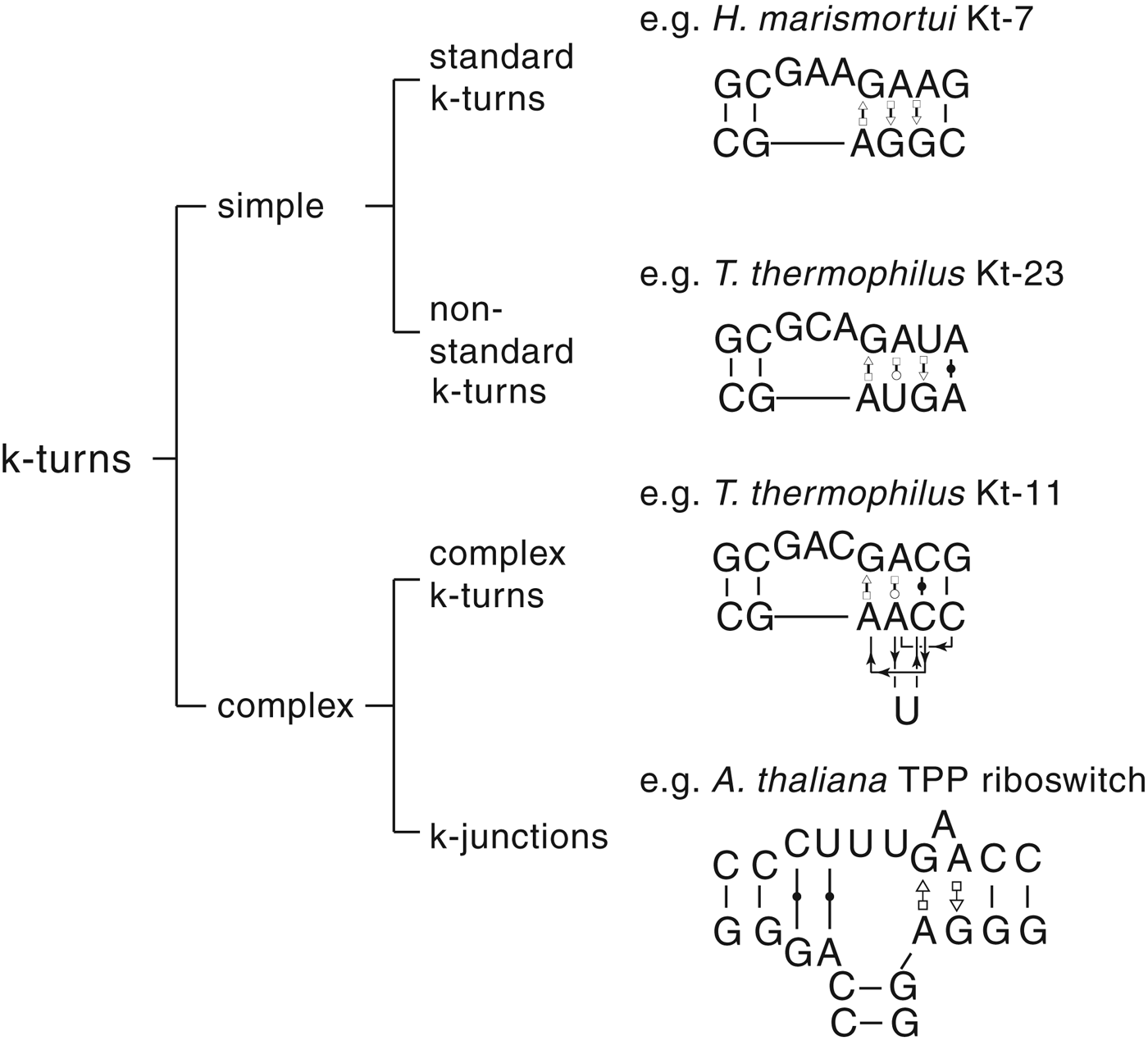
Fig. 6. A scheme showing the classification of k-turns, with examples. In the simple, non-standard k-turns one or more nucleotides of the G•A basepairs are substituted by a different nucleotide. In the complex k-turns the nucleotides of the tandem G•A basepairs do not map linearly onto the sequence. k-junctions contain an additional helix on the non-loop-containing strand.
6.1 Non-standard, simple k-turns
In non-standard k-turns, there is a substitution of one of the nucleotides in the otherwise-highly conserved tandem G:A, A:G pairs at the 1b:1n or 2b:2n positions. This occurs most frequently at the 2b:2n position. Kt-23 of the 16S rRNA in the small ribosomal subunit provides a good example. In this k-turn the 2n nucleotide is rather infrequently found to be G, with a frequency of occurrence U > C > G > A. While a G2nU substitution in HmKt-7 prevented ion-induced folding in vitro, Kt-23 from Thermus thermophilus (where 2n = U) folded normally into the kinked conformation on the addition of Mg2+ ions. Moreover, in the T. thermophilus ribosome Kt-23 forms a completely normal k-turn structure (Fig. 7), that superimposes with HmKt-7 with an root mean square deviation (RMSD) = 0·60 Å. The A:G pair at the 2b:2n position is replaced by a trans Hoogsteen-Watson Crick A•U pair, that places the adenine nucleobase in essentially the location that it would be found in a standard k-turn. The A1n is also in its normal position, and both adenine nucleobases accept the standard cross-strand H-bonds. The k-turn adopts an N1 conformation structure, with an H-bond from G-1n O2′ to A2b N1 plus G-1n N2 to A2b N3, and GL1 O2′ to A1n N1. The 3b:3n base pair is a trans U:G basepair with a C1′-C1′ distance of 11·0 Å; the local widening of the helix may be important, see section 5.

Fig. 7. The structure of T. thermophilus Kt-23, a simple, non-standard k-turn with U at the 2n position in place of the normal G. The sequence is shown (upper) with the non-standard U2n nucleotide highlighted in cyan. A superposition of TtKt-23 (colored; PDB ID 2WH1) with HmKt-7 (grey) is shown as a parallel-eye stereoscopic image (lower). This shows the close superposition of the key A1n, A2b and G-1n nucleotides between the two structures despite the G2nU substitution, making all the standard cross-strand H-bonds.
The least-common 2b:2n sequence found in Kt-23 is A:A at <1%. Bioinformatic searching revealed an example that occurs in Telohania solenopsae (Schroeder et al. Reference Schroeder, Daldrop, Mcphee and Lilley2012), a parasite of the fire ant, Solenopsis invicta. It is a simple k-turn that only differs from a standard k-turn in the 2n position. It exhibits weak folding in Mg2+ ions but folds normally on binding L7Ae protein. It was functional on substitution into the SAM-I riboswitch in place of the normal k-turn, i.e. the modified riboswitch bound SAM normally. The structure of T. solenopsae k-turn was solved in the context of the riboswitch by crystallography (Schroeder et al. Reference Schroeder, Daldrop, Mcphee and Lilley2012). It adopted the standard k-turn conformation, with all the normal H-bonds (Fig. 8). It could be superimposed with HmKt-7 with an RMSD of 1·6 Å and an almost perfect superposition of the A1n and A2b nucleobases. These accept the normal cross-strand H-bonds, forming an N1 structure. The A2b:A2n pairing is disposed like a trans-Hoogsteen-sugar edge basepair, although the nucleobases are spaced apart (e.g. A2b N6 - A2n N3 = 5·0 Å) and unconnected by H-bonds.
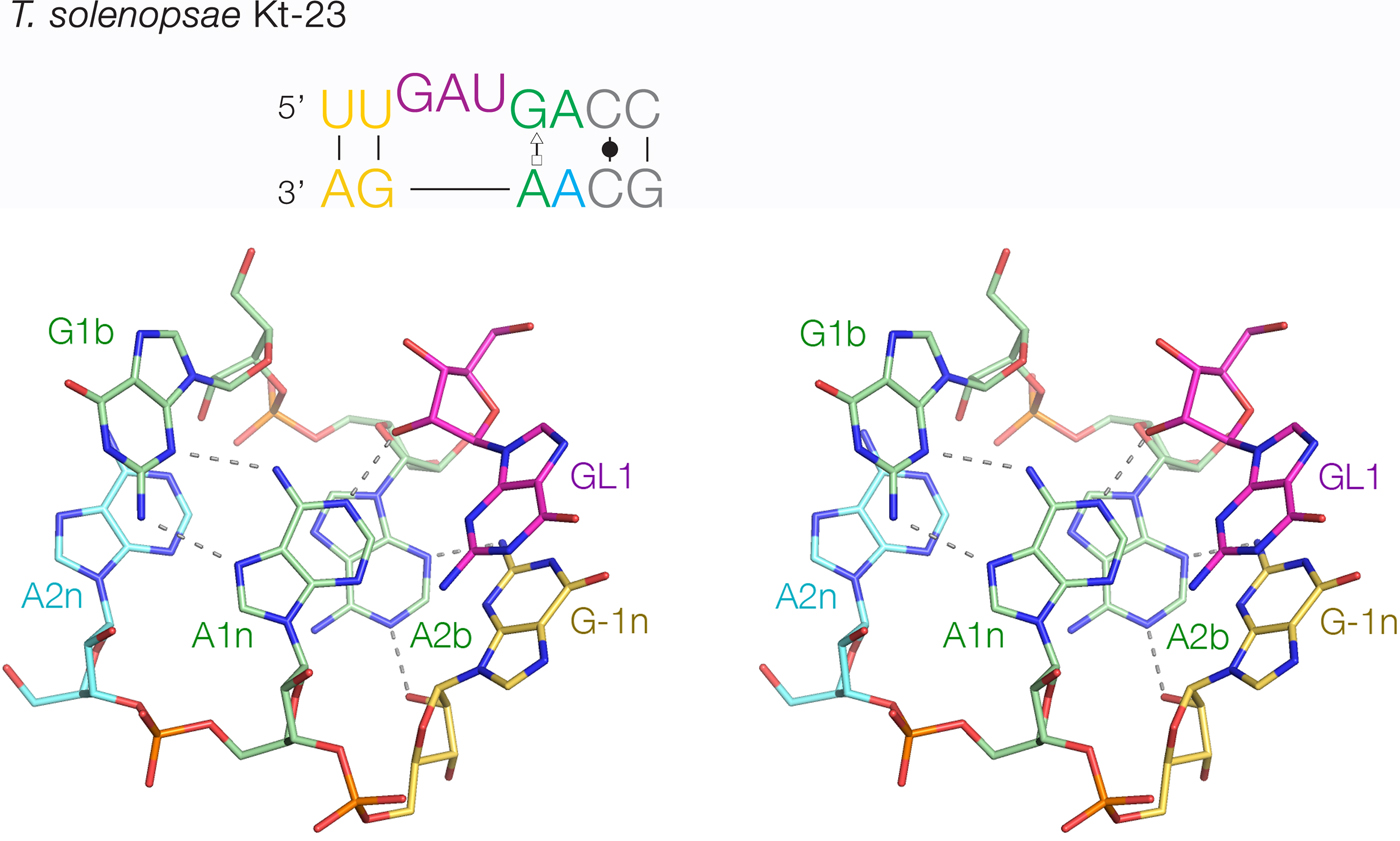
Fig. 8. The structure of T. solenopsae Kt-23, a simple, non-standard k-turn with A at the 2n position in place of the normal G (Schroeder et al. Reference Schroeder, Daldrop, Mcphee and Lilley2012). The sequence is shown (upper) with the non-standard A2n nucleotide highlighted in cyan. The core of the TsKt-23 is shown as a parallel-eye stereoscopic image (lower; PDB ID 4AOB), showing that all the standard cross-strand H-bonds are present.
6.2 Complex k-turns
In complex k-turns the nucleotides of the tandem G•A basepairs do not map onto the sequence in a linear way – they appear out of place in the normal secondary structure depiction. However, on folding into the k-turn conformation they nevertheless find their conventional position within the structure and make the same interactions. We, therefore, use our standard nomenclature to label these nucleotides according to their position in the structure, rather than the sequence.
Kt-11 in the small ribosomal subunit of T. thermophilus provides a good example (Fig. 9). In the NC helix, the non-bulged strand has the linear order 5′–C4n–A2n–U–C3n–A1n–3′. The backbone forms an S-shaped turn that places C3n under A2n despite being to its 3′ side in the sequence, and the 1n and 2n nucleotides are separated by two nucleotides in the primary sequence. Despite the scrambled connectivity of this sequence, the RNA folds into a normal k-turn structure in the ribosome with all the nucleotides in their correct places. Once again, the A1n and A2b are placed in the normal k-turn positions in the structure, with the near-universal cross-strand H-bonding. A2b N1 accepts a proton from G-1nO2′, so the structure is in the N1 conformation. The A2b:A2n basepair is trans-Watson-Crick-Hoogsteen, connected by two H-bonds, with a significant rotation of the A2n nucleobase relative to the normal G2n in HmKt-7 for example. Interestingly, the structure of Kt-11 in the Escherichia coli ribosome is virtually identical to that of T. thermophilus. The non-bulged strand of the NC helix makes a pronounced S-turn with the linear order 5′−G4n−A2n−U−U3n−A1n−3′, and the A2b:A2n basepair is trans-Hoogsteen-Watson-Crick. Nevertheless, the RNA again folds into the k-turn geometry, placing A1n and A2b into their standard positions where they make the usual cross-strand H-bonds in the N1 conformation. The core can be superimposed with that of HmKt-7 (Fig. 9c) with an RMSD = 0·58 Å.

Fig. 9. The structure of the complex k-turn T. thermophilus Kt-11. The sequence of the k-turn is shown top left. (a). A cartoon of the structure of TtKt-11, showing the connectivity of the nucleotides, that are labeled according to their position in the k-turn structure. (b) The overall structure of TtKt-11 (PDB 2WH1) viewed from the side of the non-loop strand. (c) The core of TtKt-11 (colored) superimposed with that of HmKt-7 (grey). There is good superposition of the key A1n, A2b and G-1n nucleotides, with an RMSD = 0·579 Å. (b) and (c) are shown as parallel-eye stereoscopic images.
We noticed that a topologically-equivalent k-turn occurs in a structure determined for the Constitutive Transport Element RNA bound to the export factor TAP (Teplova et al. Reference Teplova, Wohlbold, Khin, Izaurralde and Patel2011). The non-bulged strand has the linear order 5′−U4n−A2n−A−G−A−C3n−A1n−3′, and the A2b:A2n basepair is again trans-Hoogsteen-Watson-Crick. The S-turn trajectory of the non-bulged strand is even more pronounced than TtKt-11 as it now has three consecutive nucleotides in the strand section that passes backwards, yet once again A1n and A2b are located in the correct position to make the normal cross-strand H-bonding interactions with GL1 and G-1n.
6.3 k-junctions
k-junctions represent the most extreme form of the k-turn. These went largely unnoticed for several years but were identified when a computer program was written to search for helices with the relative disposition of those in k-turns within RNA structures downloaded from the PDB (Wang et al. Reference Wang, Daldrop, Huang and Lilley2014). While this exercise identified a number of known k-turns, it surprisingly flagged the thiamine pyrophosphate (TPP) riboswitches which on closer inspection were found to contain a new class of k-turn (Wang et al. Reference Wang, Daldrop, Huang and Lilley2014). These riboswitches contain a three-way junction from which two helices emanate that interact to create the ligand binding site. But it emerged that the junction was, in fact, a k-turn, where the third helix (we call the T helix) is located on the non-bulged strand opposite the loop of the k-turn. This had also been noted in a footnote in an earlier study of A-minor interactions by Jaeger and colleagues (Geary et al. Reference Geary, Chworos and Jaeger2011).
The Arabidopsis thaliana TPP riboswitch (Thore et al. Reference Thore, Leibundgut and Ban2006) contains a good example of a k-junction (Fig. 10). As ever, the core of the k-junction has tandem G:A, A:G pairs, labeled according to our nomenclature. Both are trans sugar(G):Hoogsteen(A) basepairs. G1b and A2b are separated by an additional A that is directed down into the major groove of the NC helix to make a triple base interaction with the U5b:A5n basepair. The C helix terminates at the junction with conventional Watson Crick C:G and U:A basepairs, but for reasons that will shortly become apparent we designate the C and U as formally part of the loop, termed L1 and L1·1. These two basepairs lie between and coaxial with the C and T helices in the folded structure. L1 O2′ donates an H-bond to A1n N1 and G-1n donates H-bonds from O2′ and N2 to A2b N1 and N3 respectively. Thus the standard cross-strand H-bonds are present, and the k-junction adopts an N1 conformation. The structure can be well superimposed with a standard N1 k-turn such as ribosomal HmKt-7. Superposition using the backbone atoms for −1n, L1, 1n, 2b and 2n gives an RMSD of 1·58 Å.

Fig. 10. The structure of the A. thaliana TPP riboswitch (Thore et al. Reference Thore, Leibundgut and Ban2006) k-junction (Wang et al. Reference Wang, Daldrop, Huang and Lilley2014). The sequence of the k-junction is shown at the top. The additional helix (named T) is fused into the non-loop strand making a three-way junction with the C and NC helices. There is a bulged A (colored cyan) inserted in between G1b and A2b. The cytosine labeled L1 is basepaired with guanine, yet functionally this has the role normally taken by L1, with its O2′ is H-bonded to the A1n nucleobase. On the opposite strand, we define the next nucleotide 3′ as the −1n; its O2′ is H-bonded to the A2b nucleobase. (a) The overall structure of the k-junction viewed from the side of the loop (PDB ID 3D2G). The T helix is at the back in this view. The T and C helices are coaxial. (b) The core of the k-junction, showing the key H-bonding interactions equivalent to those of the regular k-turn structures. (a) and (b) are shown as parallel-eye stereoscopic images.
The E. coli TPP riboswitch (Serganov et al. Reference Serganov, Polonskaia, Phan, Breaker and Patel2006) also contains a k-junction in an equivalent position. The global shape is closely similar to that of the A. thaliana TPP riboswitch, with the coaxial stacking of the C and T helices. The main difference is that the 1b nucleotide is an adenine so that the 1b:1n basepair is a trans-Hoogsteen-Hoogsteen A:A basepair. What takes the role of the loop is a CGU sequence, where the CL1 and GL2 are basepaired with their complements on the opposing strand. The A1n and A2b play their usual role, accepting H-bonds L1 O2′ to A1n N1, G-1n O2′ and N2 to A2b N1 and N3, respectively. Thus this k-junction is also an N1 structure. An A1nC mutant of the riboswitch exhibited impaired folding and no longer bound TPP in calorimetric experiments (Wang et al. Reference Wang, Daldrop, Huang and Lilley2014), demonstrating the importance of the k-junction in the function of the riboswitch.
Analysis of over 11 000 TPP riboswitch sequences indicates that the k-junction is common to all and that the critical adenines at the 1n and 2b positions are conserved in >99·5% of the sequences. In a wider context, the k-junction occurs in the ribosome, with two examples in the H. marismortui 50S ribosomal subunit. Given that k-junctions are not easy to identify without detailed analysis, it is likely that further examples are waiting to be found.
We note that all the known complex k-turns and k-junctions adopt the N1 conformation. While this is more stable than the N3 conformation in about half the standard k-turns, it clearly confers an advantage in the more diverged structures. It is likely that the N1 conformation provides some structural flexibility that allows the k-turn geometry to form in the more complex sequences.
6.4 To be or not to be a k-turn
The potential of the adenine nucleobases in tandem G•A pairs for participating in A-minor interactions can be exploited more widely. In the human signal recognition particle RNA, tandem trans sugar-Hoogsteen G:A basepairs mediate a long-range tertiary interaction between two helices (Ahl et al. Reference Ahl, Keller, Schmidt and Weichenrieder2015). N3 of one adenine accepts an H-bond from the O2′ of a cytosine in the other helix, while its O2′ is H-bonded to the O2′ of the same cytosine. As we have recently discussed (Huang et al. Reference Huang, Ashraf, Wang and Lilley2017), this is strikingly similar to the interactions between A2b and the −1n nucleotide in k-turns, although there is no interaction corresponding to that between A2n N1 and the L1 O2′. However, it suggests that the definition of the k-turn might be extended further.
Conversely, the role of k-turns within a given species may be taken up by a different kind of kink in the helix, acting in a functionally-equivalent manner, but lacking all the characteristic structural features of the k-turn. In the ribonuclease P ribozyme, a tightly-kinked RNA helix (Meyer et al. Reference Meyer, Westhof and Masquida2012b) facilitates the formation of a loop–receptor interaction that creates the binding site for the substrate. In some species, this is achieved using a k-turn but in others, a different kinked structure termed the pk-turn is used. Globally the pk-turn of the Thermotoga maritima ribozyme is very similar to the geometry of a k-turn, but it lacks all the standard features of the k-turn including the G:A pairs. Yet the pk turn and the k-turn can be exchanged with preservation of function. Mutation of T. maritima RNase P to replace the pk turn by a k-turn resulted in retention of ribozyme activity, while a SAM-I riboswitch in which the k-turn had been replaced by the pk turn retained the ability to bind SAM ligand (Daldrop et al. Reference Daldrop, Masquida and Lilley2013).
Another example of structural and functional redundancy is found in the lysine riboswitch. A putative k-turn was identified in the Bacillus subtilus lysine riboswitch and shown to bind L7Ae protein (Blouin & Lafontaine, Reference Blouin and Lafontaine2007). However, a k-turn was not found in the structure of the lysine riboswitch of T. maritima (Garst et al. Reference Garst, Heroux, Rambo and Batey2008; Serganov et al. Reference Serganov, Huang and Patel2008). Rather its function was taken by a different kinked structure lacking G:A pairs, and not stabilized by A-minor H-bonds.
6.5 Identification of k-turns in RNA sequence and structure
In the light of experience studying k-turns over the last decade, how easy is it to recognize k-turn motifs within RNA structures, or indeed potential k-turns given RNA sequence information?
Identifying potential k-turns in RNA sequence reliably is not easy, and in principle, the two strands could be widely separated within the overall RNA structure. It is relatively straightforward to predict basepairing and secondary structure by analysis of nucleotide covariation, yet the k-turns can still be difficult to find. In general, we use three stages to do this, performed manually:
1. Focus on junctional sections and try to identify tandem G:A, A:G pairs, since these are the most well-conserved features. Where available, phylogenetic data should indicate strong conservation of the two adenine nucleotides.
2. Seek unpaired nucleotides (normally three) preceding the G:A 5′ to the G.
3. Check the −1b:−1n position. In the majority of k-turns, this is C:G. This is not required, but if a given sequence conforms then it adds confidence that it really is a k-turn.
While this works well for standard k-turns, it gets progressively more difficult for non-standard and complex k-turns or k-junctions. There are also procedures based on statistical methods that have been proposed for identifying k-turn motifs (Bayrak et al. Reference Bayrak, Kim and Schlick2017).
We have applied this approach to the collection of 224 candidate structured RNA sequences recently identified by Breaker and colleagues (Weinberg et al. Reference Weinberg, Lunse, Corbino, Ames, Nelson, Roth, Perkins, Sherlock and Breaker2017) by comparative analysis of specific subsets of intergenic regions. Rigorous application of the method identified four putative standard k-turns, and one k-junction. These are shown in Fig. 11 and listed in Table 2.

Fig. 11. Putative k-turns and one k-junction identified in a collection of 224 candidate structured RNA sequences from the Breaker laboratory (Weinberg et al. Reference Weinberg, Lunse, Corbino, Ames, Nelson, Roth, Perkins, Sherlock and Breaker2017). The most common sequence amongst the variations found in nature are shown for each. These k-turns and k-junction are tabulated in Table 2, with their predicted properties.
Table 2. Four putative k-turns and one k-junction within a series of 224 structured RNA sequences that have recently been presented by Breaker and colleagues (Weinberg et al. Reference Weinberg, Lunse, Corbino, Ames, Nelson, Roth, Perkins, Sherlock and Breaker2017)
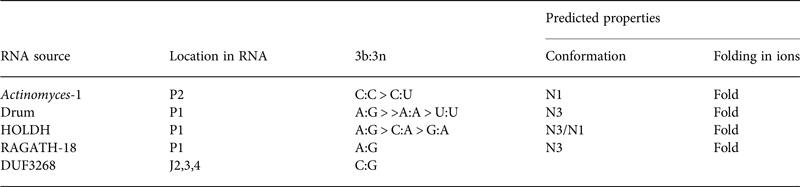
The k-turn structures were identified applying the rules written in the text. Where available, the relative frequency of the most common 3b:3n sequences are given, and the most frequent are shown in Fig. 11. The properties of the k-turns are predicted by application of the data summarized in Fig. 17. The conformations of Actinomyces-1, drum and RAGATH-18 are predicted unambiguously, but the distribution of sequences for HOLDH suggest this is more variable. We do not know how reliably the rules can be applied to a k-junction, although we note that all other known k-junctions adopt an N1 conformation.
Identifying k-turns within three-dimensional RNA structure is a rather different problem, but once again the task becomes progressively more difficult moving from standard k-turns to the more complex varieties. Two computational tools designed to analyze RNA structure include can search for k-turns. These are the RNA 3D Motif Atlas (Parlea et al. Reference Parlea, Sweeney, Hosseini-Asanjan, Zirbel and Leontis2016) and RMDetect (Cruz & Westhof, Reference Cruz and Westhof2011). Simple k-turns have generally been identified by authors publishing new structures, but more complex ones have often been missed, especially the k-junctions. We found the k-junctions by focusing on the disposition of the C and NC helices (Wang et al. Reference Wang, Daldrop, Huang and Lilley2014) (see section 6.3). A program written in Python 2·7 downloaded RNA structures from the PDB and then searched for helices with the required relative inclination similar to that of known k-turn structures. Standard Watson-Crick (plus G:U) basepairs were identified, from which four vectors were defined; one for position and three for orientation. This was calibrated against known k-turn structures and then used to search for new structures. A full description of the program was given in the Supplementary Information of Wang et al. (Reference Wang, Daldrop, Huang and Lilley2014). Application of the program identified the k-junctions of the TPP riboswitches and the Constitutive Transport Element RNA.
7. The folding of k-turns
A k-turn in free solution in a buffered solution lacking added metal ions or proteins is predominantly unfolded (Goody et al. Reference Goody, Melcher, Norman and Lilley2004), adopting the structure generally formed by an RNA duplex containing a three-nucleotide bulge (Lilley, Reference Lilley1995). The k-turn conformation must be stabilized by something specific, either the presence of metal ions, protein binding or the formation of tertiary interactions.
Experimentally folding has generally been studied mainly by two methods. One is polyacrylamide gel electrophoresis which is sensitive to the angle included by the two helical arms that have normally been extended to increase sensitivity, usually using DNA for synthetic convenience (Lilley, Reference Lilley2008). The other main approach is to use fluorescence resonance energy transfer (FRET) of RNA with terminally-attached donor and acceptor fluorophores. The efficiency of energy transfer (E FRET) depends on the inverse sixth power of the distance between the fluorophores (Förster, Reference Förster1948), and this distance becomes shorter when the RNA becomes more kinked. Small-angle X-ray scattering interferometry with RNA containing terminally-attached gold nanoparticles has also been used (Shi et al. Reference Shi, Huang, Lilley, Harbury and Herschlag2016), where Fourier transformation of the processed scattering curves yields the distance between the nanoparticles. EPR has also been used to study the structure of k-turns (Esquiaqui et al. Reference Esquiaqui, Sherman, Ye and Fanucci2016).
Fluorescence lifetime distributions of donor-acceptor-labeled k-turn-containing RNA indicated that in the absence of added metal ions and proteins there is a conformational equilibrium between folded (i.e. kinked) and unfolded (extended) RNA that is strongly biased towards the unfolded state (Goody et al. Reference Goody, Melcher, Norman and Lilley2004). We have found three main mechanisms that affects the position of this equilibrium, driving the population into the kinked conformation.
7.1 Folding of k-turns on the addition of metal ions
Some k-turns (although not all, as we discuss below) will adopt the folded conformation on the addition of metal ions (Goody et al. Reference Goody, Melcher, Norman and Lilley2004; Liu & Lilley, Reference Liu and Lilley2007). This is revealed as a retardation of electrophoretic mobility, or an increase in FRET efficiency E FRET. Titration curves of E FRET as a function of ionic concentration are well fitted by a two-state folding process (Fig. 12a). Folding occurs in either divalent or monovalent ions, with half concentrations of [Mg2+]1/2~100 µM and [Na+]1/2~30 mM. The intracellular concentration of unbound Mg2+ ions has been measured using fluorescent indicator probes at close to 1 mM (Csernoch et al. Reference Csernoch, Bernengo, Szentesi and Jacquemond1998; Li-Smerin et al. Reference Li-Smerin, Levitan and Johnson2001), so this would be sufficient to achieve folding.

Fig. 12. Folding of HmKt-7 in response to the addition of metal ions and the binding of protein. In these experiments, the folding of a 27 bp RNA duplex with a centrally-located HmKt-7 is studied by fluorescence spectroscopy (top). The RNA is 5′-terminally labeled with fluorescein and Cy-3, and the efficiency of energy transfer between the fluorophores (E FRET) measured as a function of the ion or protein concentration. In the absence of these, the structure is relatively extended with a corresponding low value of E FRET. On folding into the kinked conformation the distance between the fluorophores shortens, resulting in an increase in E FRET (these species are shown schematically in part (a). As the population of the kinked form increases the mean value of E FRET rises, and the data are well fitted by a two-state model, shown by the lines in the plots. (a) Titration with Mg2+ ions. The fit corresponds to two-state folding induced by the non-cooperative binding of Mg2+ ions with a [Mg2+]1/2 ~ 100 µM. (b) Titration with AfL7Ae protein. Addition of the protein leads to a folding of the k-turn, with a typically slightly higher end point compared with the ion titration. The affinity of L7Ae for k-turn RNA is extremely high (Turner & Lilley, Reference Turner and Lilley2008), so that binding is essentially stoichiometric and cannot be used to estimate an apparent K d.
7.2 Folding of k-turns on the binding of proteins
Many k-turns in nature are bound by specific proteins. The archetypal k-turn binding proteins are the L7Ae family (discussed below). The folding of HmKt-7 induced by the binding of Archeoglobus fulgidus L7Ae (AfL7Ae) is shown using FRET in Fig. 12b. The affinity of L7Ae for k-turn RNA is extremely high, and the folding observed as the increase in E FRET reflects stoichiometric binding as discussed in section 9. Protein-induced folding is not restricted to proteins of the L7Ae family. The majority of k-turns in the ribosome are bound by proteins of greatly differing structure and recognized in very different ways. This can be seen by the examples of E. coli L24 and T. thermophilus S17 (Fig. 13a, c). FRET analysis shows that Kt-7 adopts the kinked conformation on the binding of either protein (Fig. 13b, d). Thus the folding of k-turn RNA on binding proteins in general, is not a property of a single class of proteins.
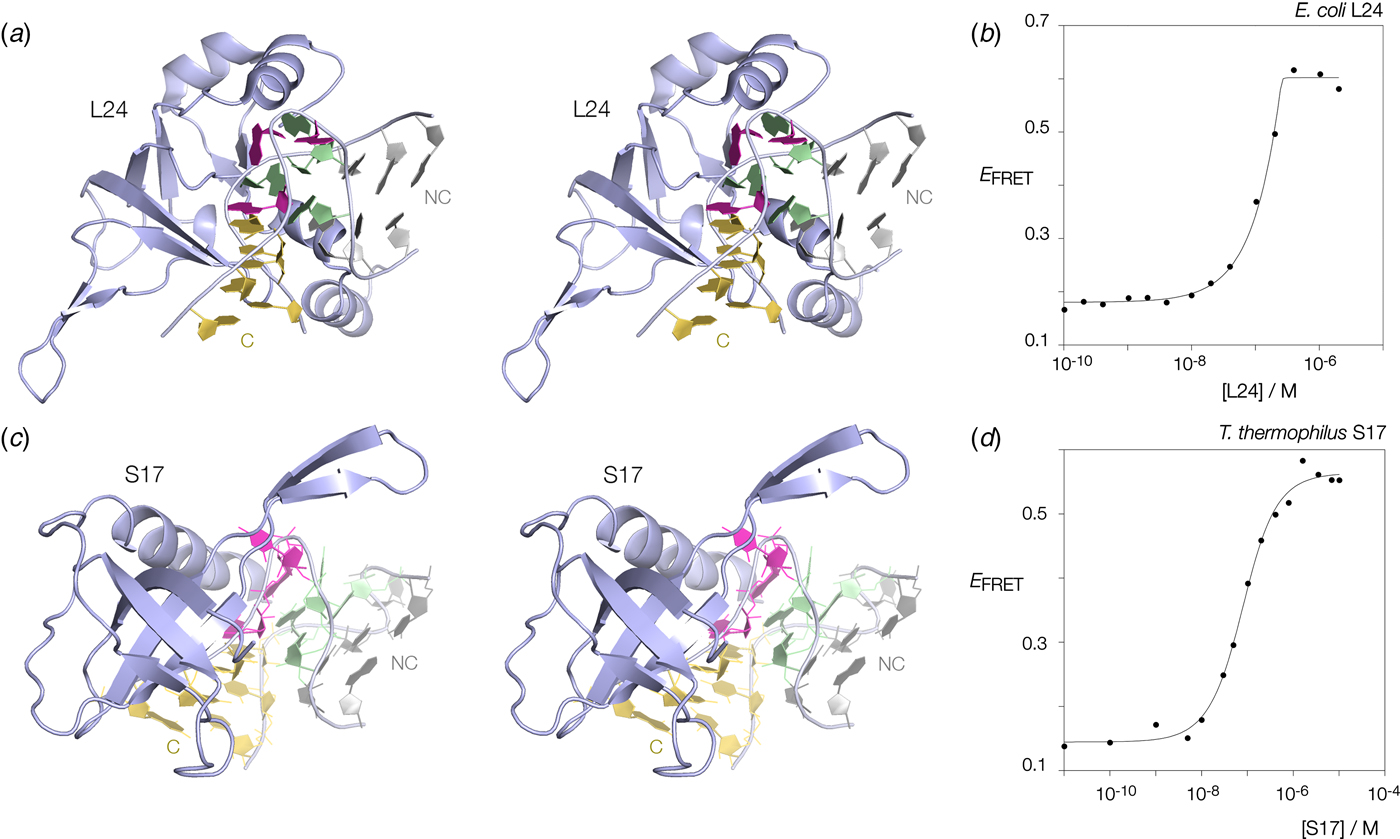
Fig. 13. Protein-induced folding of k-turns by ribosomal proteins other than L7Ae. Two examples are shown here, one from the large and one from the small ribosomal subunit. (a) Parallel-eye stereoscopic image of L24 bound to Kt-7 in the H. marismortui 50S ribosomal subunit, PDB ID 2QA4 (Kavran & Steitz, Reference Kavran and Steitz2007). The core of the protein comprises 9 strands of β sheet that organize peripheral α helices that interact with the RNA. Two α helices at the N- (17–22) and C- (106–113) terminal ends of the protein define a cleft through which the non-loop strand of the k-turn passes. The N-terminal α helix is quite basic and lies adjacent to the minor groove, making only non-specific contacts. The C-terminal α helix is directed into the major groove and is somewhat equivalent to the helix of the L7Ae proteins (see Fig. 14). Asp 105 makes two H-bonds with G1b, and the nucleobase lies at the N-terminal pole of the α helix dipole. There is no contact with the loop nucleotides of the k-turn. (b) Folding of HmKt-7 induced by the binding of L24 protein analysed by FRET. The experiment was performed analogously to that in Fig. 12b. E FRET rises upon addition of the L24 protein as the RNA population is driven into the folded conformation of the k-turn. The line is a fit to a two-state model of folding induced by protein binding. (c). Parallel-eye stereoscopic image of S17 bound to Kt-11 in the T. thermophilus 30S ribosomal subunit, PDB ID 4V5E (Weixlbaumer et al. Reference Weixlbaumer, Jin, Neubauer, Voorhees, Petry, Kelley and Ramakrishnan2008). The structure of S17 is very different from either L24 or L7Ae, comprising 7 strands of β sheet plus coil, and a single C-terminal α helix. The loop of the k-turn lies between the α helix and the β domain. β strand 40–45 plus the coil region 12–16 make backbone contacts with the loop strand of the C helix, while the α helix passes across the apex of the k-turn in a transverse manner, above the L1 and L2 nucleobases. Tyr 95 makes a hydrophobic interaction with these. (d) Folding of HmKt-7 induced by the binding of S17 protein analysed by FRET. In a similar manner to L24, E FRET rises upon addition of the S17 protein as the RNA population is driven into the folded conformation of the k-turn. The line is a fit to a two-state model of folding induced by protein binding.

Fig. 14. The molecular interaction between an archaeal L7Ae protein and the standard k-turn HmKt-7.
The complex between L7Ae of A. fulgidus and HmKt-7 as a stem-loop structure was solved at 2·3 Å resolution (Huang & Lilley, Reference Huang and Lilley2013). Parallel-eye stereoscopic images of the structure (PDB 4BW0) are shown. (a) An overall view of the complex. The L7Ae protein is shown in cartoon form, with the sections making contact with the RNA highlighted in blue. The view is from the non-loop side of the k-turn so that the C helix is directed rightwards. (b) The alpha helix located in the major groove on the outer face of the k-turn. Key amino acid side chains that interact with the RNA are shown in stick form. K37 and R41 contact the backbone while N33 and E34 make specific interactions with the guanine nucleobases of the G:A basepairs G2n and G1b, respectively. (c) The hydrophobic loop that caps the loop region of the k-turn. Note the hydrophobic side chains of I88 and V90 on the face of the protein loop contacting the L1 and L2 nucleobases. In this complex, the carboxylate group of E89 makes a specific contact with the GL1 nucleobase.
7.3 Folding of k-turns induced by tertiary interactions
Tertiary interactions can also stabilize the folded conformation of the k-turn. This was first shown in a G2nA mutant of the SAM-I riboswitch k-turn (Schroeder et al. Reference Schroeder, Daldrop and Lilley2011). Although this k-turn was unable to fold in isolation on the addition of metal ions, it did fold in the context of the riboswitch. A crystallographic structure of the RNA showed all the normal interactions of the k-turn despite the presence of the mutant A2b:A2n basepair. In the riboswitch the C-helix mediates a tertiary contact between its terminal loop and a receptor, creating the SAM ligand binding site (demonstrated by calorimetry), and evidently, these tertiary contacts lead to the induced folding of the k-turn.
8. Recognition of k-turns by proteins of the L7Ae family
The L7Ae family proteins are found bound to a variety of k-turns in different cellular structures (Koonin et al. Reference Koonin, Bork and Sander1994; Watkins et al. Reference Watkins, Segault, Charpentier, Nottrott, Fabrizio, Bachi, Wilm, Rosbash, Branlant and Luhrmann2000). The family includes the archaeal and eukaryotic proteins L7Ae, L30e and S12e, the human 15·5k protein (Nottrott et al. Reference Nottrott, Hartmuth, Fabrizio, Urlaub, Vidovic, Ficner and Luhrmann1999) and the yeast Nhp2 and Snu13p proteins. Functional substitutions between some of these proteins have been found to be tolerated (Rozhdestvensky et al. Reference Rozhdestvensky, Tang, Tchirkova, Brosius, Bachellerie and Hüttenhofer2003). YbxF and YlxQ are bacterial homologs (Baird et al. Reference Baird, Zhang, Hamma and Ferré-D'amaré2012). L7Ae-family proteins are bound to k-turns in the ribosome (Ban et al. Reference Ban, Nissen, Hansen, Moore and Steitz2000), the U4-U6.U5 spliceosomal complex (Nottrott et al. Reference Nottrott, Hartmuth, Fabrizio, Urlaub, Vidovic, Ficner and Luhrmann1999; Vidovic et al. Reference Vidovic, Nottrott, Hartmuth, Luhrmann and Ficner2000), box C/D (Kuhn et al. Reference Kuhn, Tran and Maxwell2002; Szewczak et al. Reference Szewczak, Degregorio, Strobel and Steitz2002; Watkins et al. Reference Watkins, Dickmanns and Luhrmann2002) and H/ACA snoRNPs (Hamma & Ferré-D'Amaré, Reference Hamma and Ferré-D'amaré2004; Li & Ye, Reference Li and Ye2006; Rozhdestvensky et al. Reference Rozhdestvensky, Tang, Tchirkova, Brosius, Bachellerie and Hüttenhofer2003), U3 snoRNP (Marmier-Gourrier et al. Reference Marmier-Gourrier, Clery, Senty-Segault, Charpentier, Schlotter, Leclerc, Fournier and Branlant2003), telomerase (Pogacic et al. Reference Pogacic, Dragon and Filipowicz2000) and archael RNaseP (Cho et al. Reference Cho, Lai, Susanti, Mukhopadhyay and Gopalan2010). L7Ae-family proteins are thus very much the archetypal k-turn binding proteins, essential in many cellular processes including ribosome structure, spliceosome assembly and guided site-specific modification of RNA. RIP-seq experiments have indicated that translation of L7Ae is autoregulated in archaea (Daume et al. Reference Daume, Uhl, Backofen and Randau2017).
Crystal structures have been determined for a number of L7Ae-family protein-RNA complexes. These include 15·5k bound to the human U4 snRNA k-turn (Vidovic et al. Reference Vidovic, Nottrott, Hartmuth, Luhrmann and Ficner2000), archaeal L7Ae bound to a box C/D k-turn (Moore et al. Reference Moore, Zhang, Fenley and Li2004; Suryadi et al. Reference Suryadi, Tran, Maxwell and Brown2005) and to a box H/ACA k-loop (Hamma & Ferré-D'Amaré, Reference Hamma and Ferré-D'amaré2004). The structure of L7Ae bound to Kt-15 has also been determined in the context of the complete large ribosomal subunit (Ban et al. Reference Ban, Nissen, Hansen, Moore and Steitz2000); however, Kt-15 is a complex k-turn, so less directly comparable with the simple k-turn complexes. We determined the structure of A. fulgidus L7Ae protein (Af L7Ae) bound to the archetypal k-turn H. marismortui Kt-7, probably the best-characterized k-turn (Fig. 14). This was solved at a near-atomic resolution of 2·3 Å, giving an electron density map of excellent quality where each side chain was unambiguous. Taking all these structures together we can describe the manner of recognition of and interaction with standard k-turns in general terms:
1. L7Ae proteins bind in the major groove that is splayed around the outside of the k-turn (Fig. 14a). While DNA binding proteins frequently place a recognition helix in the major groove to make sequence–specific interactions, this is generally not possible in RNA due to the narrow and deep geometry. However, in the k-turn the major groove is opened by its location on the outside face. Thus the L7Ae-family complexes resemble helix-turn-helix and zinc finger proteins in this respect.
2. Two elements of the protein comprise the RNA binding interface. The first is a very basic β-strand : turn : α-helix element. The second is a short loop of hydrophobic residues that make a 180° turn.
3. The α-helix is located in the major groove of the NC helix, directing its N-terminal end towards the major groove edges of the guanine nucleobases of the G:A pairs (Fig. 14b).
4. The C-terminal end of the α-helix lies close to the non-bulge-containing strand of the NC arm of the k-turn, from which basic side chains make non-specific interactions with the RNA backbone. The arginine in the third turn (R41 in Af L7Ae) makes a universal H-bond with the 3n/4n phosphate. This interaction is observed in all the complexes studied. The lysine in the second turn (K37 in Af L7Ae) is bonded to the 4n/5n phosphate in the Kt-7 complex but is more variable over the ensemble of structures. These interactions will locate the position of the NC helix, and therefore interrogate the angle of the helices included in the k-turn. At the N-terminal end of the β-sheet two additional basic side chains contribute to the non-specific electrostatic stabilization of the complex.
5. Side chains located at the N-terminal end of the α-helix make sequence–specific interactions with the guanine nucleobases of the tandem G:A, A:G pairs in the core of the k-turn. A glutamate from the first turn (E34 in AfL7Ae) is H-bonded to G1b N1. This seems to be a universal interaction, that is observed in the L7Ae-box C/D and 15·5 k-U4 k-turn interactions. The adjacent asparagine (N33 in AfL7Ae) is H-bonded to G2n O6; this also occurs in the box C/D interaction. In an equivalent complex of L7Ae with the non-standard k-turn Kt-23 of Telohenia solenopsae (where 2n = A) N33 is H-bonded to A2n N6 (Huang & Lilley, Reference Huang and Lilley2014).
6. In all the structures, G1b O6 is located almost on the axis of the α-helix at the N-terminal end. The exocyclic oxygen atom has a significant partial negative charge, that will be stabilized electrostatically by the positive pole of the helix dipole.
7. The hydrophobic loop covers the L1 and L2 nucleobases, burying a surface area of 732 Å2 in the L7Ae-Kt-7 complex and making good Van der Waals contact (Fig. 14c). Contact with the loop is maximized by the syn conformation of L2. The glutamate of the loop (E89 in A. fulgidus L7Ae) is H-bonded to L1 GN1. A similar interaction was observed in the L7Ae- box C/D complex.
The L7Ae family of proteins is highly specific for binding k-turns in RNA, and the structural analysis shows the basis for this. By making contacts in the groove of the NC helix (the helix) and the loop region (the hydrophobic loop) the protein is sensitive to the angle of the kink in the RNA. At the same time, the N-terminal region makes base-specific contacts with the two guanine nucleobases of the tandem G:A pairs. However, L7Ae makes no contacts with the C helix, so that k-loops (where the C-helix is replaced by a terminal loop) can also form complexes (Hamma & Ferré-D'Amaré, Reference Hamma and Ferré-D'amaré2004).
9. The process of binding k-turns by proteins of the L7Ae family
Binding of L7Ae proteins to k-turns result in the formation of the kinked conformation. That is true for the majority of k-turns, including many of those that will not fold in response to the addition of metal ions. There is, however, some sequence specificity, residing largely in the sequence of the loop of the k-turn, but the rules have not been fully investigated. The folding is readily followed via the change of end-to-end distance using FRET (e.g. Fig. 12b). Such data are fitted well by a two-state process. However, the data are characteristic of stoichiometric binding, indicating a high affinity. Indeed, it turns out that the affinity of binding is too high to measure by these means. Any RNA concentration that is detectable by fluorescence is too high to be in equilibrium in the binding process. Instead we studied the binding of HmKt-7 by Af L7Ae by measuring the rates of association (k on) and dissociation (k off) (Turner & Lilley, Reference Turner and Lilley2008). The former was close to diffusion-controlled and was measured by stopped-flow. The affinity of binding was then calculated from the ratio (K d = k off/k on) and found to be K d = 10 pM.
We often loosely refer to the folding of k-turns on the addition of protein as ‘induced fit’, but this has a specific meaning that may be misleading here. In principle, the change in conformation in the RNA population could result from either active or passive action by the protein. Induced fit (Koshland, Reference Koshland1958) implies that the protein binds to the RNA in its unfolded state and somehow mechanically forces the structure to fold into the kinked structure. Alternatively, the folding of the population could be driven by conformational selection (Tsai et al. Reference Tsai, Ma, Sham, Kumar and Nussinov2001). In this mechanism, the protein would bind a pre-existing sub-fraction that is folded (in the case of k-turns we know this exists (Goody et al. Reference Goody, Melcher, Norman and Lilley2004)) and thus change the position of equilibrium for the population by mass action. Thus the key difference is whether or not the folded conformation exists prior to protein binding, and this has been extensively discussed for a variety of macromolecular interactions (Csermely et al. Reference Csermely, Palotai and Nussinov2010; Hammes et al. Reference Hammes, Chang and Oas2009; Okazaki & Takada, Reference Okazaki and Takada2008; Pitici et al. Reference Pitici, Beveridge and Baranger2002; Weikl & von Deuster, Reference Weikl and Von Deuster2008; Zhou, Reference Zhou2010). Single-molecule FRET experiments were therefore used to study the conformation of k-turn RNA at the moment of binding (Wang et al. Reference Wang, Fessl, Schroeder, Ouellet, Liu, Freeman and Lilley2012). This was achieved by tethering a U1A-AfL7Ae fusion protein to a quartz slide, and then injecting fluorescent donor-acceptor-labeled HmKt-7. The fluorescent RNA becomes immobilized on the binding, and the FRET efficiency reports on the RNA conformation at a given moment.
It was observed that the bound molecules existed only in a high-E FRET state (i.e. the k-turns were folded into the kinked conformation) and remained stable so with no transitions to a low-E FRET state observed in many molecules analyzed. Real-time analysis of binding was used to try to catch bound RNA in a less-than-fully folded state, but none was detected down to an 8 ms frame rate (Fig. 15) (Wang et al. Reference Wang, Fessl, Schroeder, Ouellet, Liu, Freeman and Lilley2012). We cannot exclude the possibility that the structure is induced to form by L7Ae protein on a faster timescale. A prerequisite of the conformational selection mechanism is that the folded structures of the free and bound RNA are the same. We have determined the structure of HmKt-7 both as free RNA and bound by AfL7Ae at high resolution and found that the two states are closely similar, with an all-atom RMSD of 0·83 Å. Thus very little conformational adjustment of the k-turn structure should be required on binding to L7Ae. All the present data are fully consistent with a conformational selection mechanism, although we cannot say this is completely proven at this time.
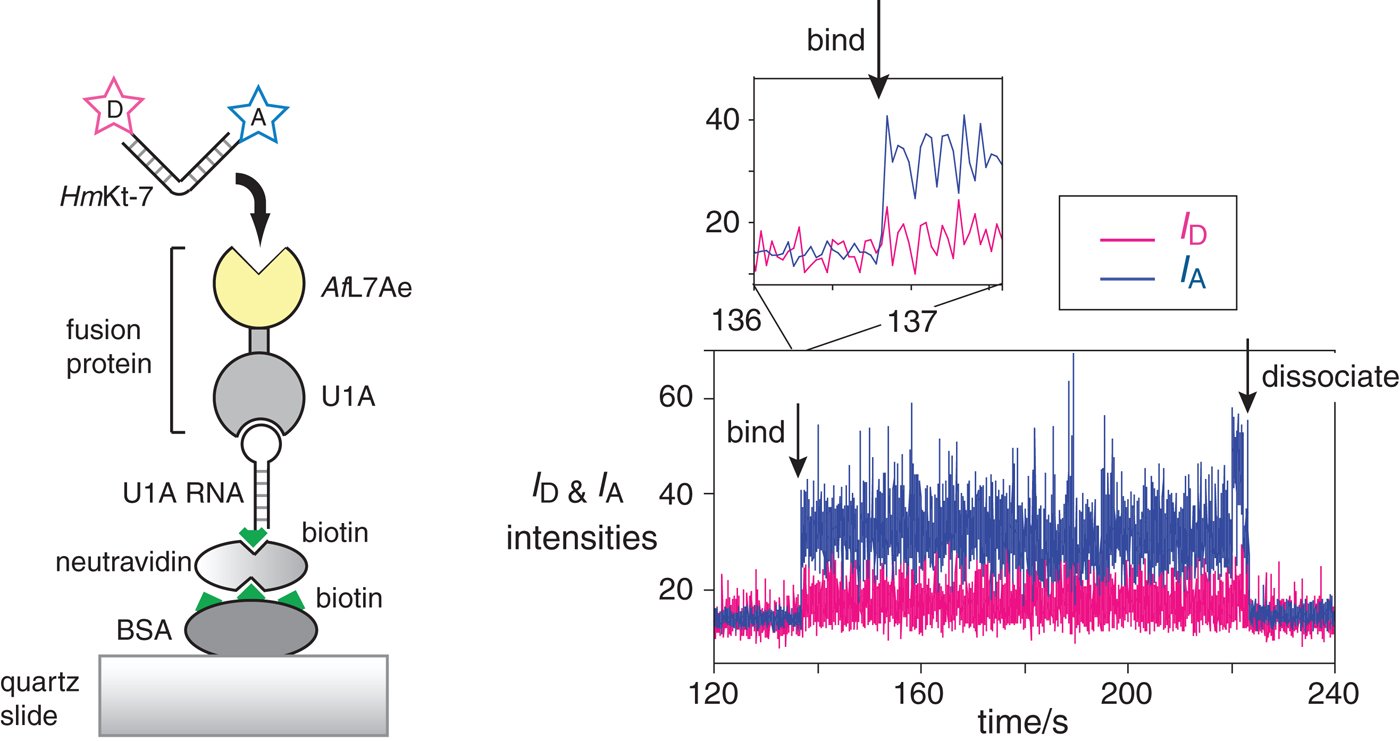
Fig. 15. Single-molecule analysis of L7Ae-induced folding (Wang et al. Reference Wang, Fessl, Schroeder, Ouellet, Liu, Freeman and Lilley2012). The scheme (left) shows the construct used. AfL7Ae (highlighted yellow) was fused at the C-terminus of U1A protein. The fusion U1A-L7Ae fusion was bound to a U1A RNA stem-loop biotinylated at its 5′ end, that was attached to the surface of a quartz slide via neutravidin-biotin-BSA. The HmKt-7 RNA was 5′-terminally-labeled with Cy3 donor and Cy5 acceptor and becomes immobilized when it binds to the AfL7Ae of the fusion. This only appears as a fluorescent spot on the image of the surface when bound. The plots of the donor (I D magenta) and acceptor (I A blue) intensity as a function of time (right) show the moments at which a single RNA molecule binds and dissociates. Expansion of the region in which RNA binds to the protein (shown as the upper trace) reveals that the bound molecule achieves full FRET efficiency within a single frame (i.e. 16 ms), consistent with it being folded at the moment of binding.
10. Sequence dependence of ion-dependent folding
We have already noted that k-turns such as HmKt-7 fold into their kinked conformation on the addition of micromolar concentrations of divalent metal ions (see Fig. 12a), or millimolar concentrations of monovalent metal ions. However, we had also found that some other k-turns, exemplified by the AfboxC/D and human U4 k-turns remain unfolded in the presence of these ions (McPhee et al. Reference Mcphee, Huang and Lilley2014). This does not result from a fundamental inability to adopt the k-turn conformation because all of these sequences fold into the standard k-turn structure on binding L7Ae proteins. Yet some k-turns fold on the addition of metal ions and some do not. A difference in some critical sequence must determine the different folding characteristics, so what is this? In fact, the possible location of such a key element is rather limited. Comparing the sequences of HmKt-7 and human U4 k-turns the tandem G:A pairs are standard, and both have −1b:−1n = C:G. We, therefore, turned to the next position in the NC helix and examined the effect of varying the 3b:3n sequence, which varies widely between k-turn sequences.
10.1 The 3b:3n sequence is a critical determinant of folding properties in metal ions
The role of the 3b:3n sequence was systematically investigated in the context of the HmKt-7 sequence by construction all possible 16 variants in that position. Their folding in response to the addition of Mg2+ ions was studied by FRET (McPhee et al. Reference Mcphee, Huang and Lilley2014). The result was unambiguous, changing the 3b:3n sequence determined the folding properties of the HmKt-7 variants between full folding (e.g. the natural sequence 3b:3n = A:G) and none at all (e.g. 3b:3n = G:C or C:G). Some underwent intermediate folding in response to the addition of metal ions (e.g. 3b:3n = A:C, or U:U) and the whole range of folding end-points was obtained for the set of k-turn variants. Yet any could be folded by the binding of L7Ae protein. All can thus adopt the k-turn conformation, but some will not do so on the addition of metal ions alone.
The dependence of the ion-dependent folding characteristics of the HmKt-7 variants can be displayed as a 4 × 4 array, where the rows are the 3b variants and the columns are the 3n variants (Fig. 16a). These have been color-coded to reflect folding ability (red = good folding and blue = poor folding in metal ions). From this, a pattern emerges, and we can define a set of ‘rules’ (McPhee et al. Reference Mcphee, Huang and Lilley2014).
1. 3b:3n = Watson-Crick base pairs (on the ascending diagonal) lead to poor or non-existent folding of HmKt-7 in metal ions. This is worst for the G:C and C:G pairs. G:U is also a very weakly-folding sequence.

Fig. 16. The effect of the 3b:3n sequence on the folding of HmKt-7 in metal ions. (a) 16 HmKt-7 variants with all possible 3b:3n sequence were synthesized as fluorophore-labeled duplexes and E FRET measured as a function of Mg2+ ion concentration (as in Fig. 12a). The end point of the titration is shown for each sequence, as a measure of the extent of folding in metal ions. These are shown in the form of a 4 × 4 array, where the rows are the 3b and the columns the 3n sequences. Those that fold well (final E FRET ⩾ 0·5) are colored red, and those that fold poorly or not at all (final E FRET ⩽ 0·3) are colored blue. (b) Close-up view of metal ions directly bound to G2n and G3n (McPhee et al. Reference Mcphee, Huang and Lilley2014). Parallel-eye stereoscopic image of a high-resolution crystal structure of HmKt-7 (PDB ID 4CS1) showing two hydrated metal ions in the major groove bound to G2n and G3n O6 atoms. The metal ions (magenta) and oxygen atoms (red) of water molecules of hydration and guanine O6 are shown as spheres. Direct metal ion–oxygen interactions are shown by broken lines. The electron density is taken from the Fo-Fc omit map contoured at 2 σ.
2. k-turns with 3b = C (second row) fold well in response to metal ions.
3. k-turns with 3n = G (third column) also fold well in response to metal ions.
4. Rule 1 takes precedence over 2 and 3 because 3b:3n = C:G fails to fold on the addition of metal ions alone.

Note that these rules are sufficient to define the folding properties of the entire folding space. Although the rules are deduced from the analysis of the folding properties of the HmKt-7 variants, they are clearly quite general. We were able to switch the folding characteristics of the human U4 k-turn from no folding to full folding in metal ions by changing the 3b:3n sequence from G:C to A:G (McPhee et al. Reference Mcphee, Huang and Lilley2014), moving from a blue to a red square in Fig. 16a in a kind of ‘snakes and ladders’ move. And we had noted a strong tendency for SAM-I riboswitch k-turns to have 3b:3n sequences that confer ion-dependent folding, while U4 k-turns have a marked propensity to have 3b:3n sequences that disallow folding in metal ions. This suggests that the rules deduced for HmKt-7 apply widely. As we shall see in section 11 below, these rules, plus those determining k-turn conformation, can rationalize a large body of data and have the considerable predictive ability.
10.2 A molecular explanation for the 3n = G folding rule
While the rules for the effect of the 3b:3n sequence on folding of k-turns in metal ions are largely empirical, we can explain the 3n = G rule. In the course of the refinement of a 2·0 Å resolution crystal structure of an RNA duplex containing HmKt-7, we found electron density in the major groove adjacent to the Hoogsteen edges of the G2n and G3n nucleobases (McPhee et al. Reference Mcphee, Huang and Lilley2014). Further refinement revealed that the density corresponded to two octahedrally-coordinated hydrated metal ions (Fig. 16b). Given the symmetry, these were probably Mg2+ ions, although the water O-metal distances were slightly ambiguous. Importantly both ions were directly coordinated to the O6 atoms of G2n and G3n as inner-sphere complexes, and indeed one of the metal ions was coordinated to the O6 atoms of both G2n and G3n. Atomic mutagenesis experiments studying the ion-dependent folding of HmKt-7 in which a given GO6 was selectively removed (i.e. guanine to 2-aminopurine substitution) revealed that both G2n and G3n O6 atoms were required for full folding in metal ions, but that of G1b was of little significance (McPhee et al. Reference Mcphee, Huang and Lilley2014). The crystallographic and atomic mutagenesis data show the importance of G3n O6 on the coordination of metal ions and k-turn folding, thus explaining the 3n = G rule.
10.3 The −1b:−1n sequence is another important determinant of folding properties in metal ions
For the great majority of ribosomal and riboswitch k-turns −1b:−1n = C:G, and in our original analysis of the sequence dependence of k-turn folding properties we ignored this position, so it remained constant as C:G in all the constructs. However, we were aware that an important k-turn did not conform to the rules discussed in the previous section, the Afbox C/D k-turn. This has 3b:3n = U:U that in the HmKt-7 context confers reasonably good folding in metal ions, yet the Afbox C/D k-turn exhibits no folding in metal ions alone (although it folds on binding AfL7Ae). We, therefore, systematically exchanged four elements between the Afbox C/D and HmKt-7 k-turns, i.e. the −1b:−1n, 3b:3n, 4b:4n and loop sequences, and examined the folding properties in metal ions (Ashraf et al. Reference Ashraf, Huang and Lilley2017). From this analysis, we deduced that the most important difference between this two k-turn is the −1b:−1n sequence, which is G:C in the Afbox C/D k-turn. Just inverting this basepair in HmKt-7 (i.e. converting to −1b:−1n = G:C) leads to very poor folding in the presence of metal ions. The reverse change in the Afbox C/D k-turn (generating −1b:−1n = C:G) leads to the largest improvement in ion-dependent folding for a single change. While the 4b:4n sequence had a small effect on folding characteristics, this was smaller and less systematic than that for −1b:−1n. Probably, for this reason, the great majority of k-turns have evolved −1b:−1n = C:G. This includes complex k-turns such as HmKt-11, and the TPP riboswitch k-junctions. The biggest exception to this is the box C/D k-turns, that may be in part explained by a requirement to create an adenine N6-methylation target in some of these (see section 12).
11. The distribution of sequences for natural k-turn elements
We have now seen that the 3b:3n base pair plays the key role in determining both the conformation of the k-turn (i.e. N3 or N1, Fig. 4), and the folding properties of the k-turn, whether or not it will fold in response to metal ions alone (Fig. 16). These empirical rules are combined and summarized in Fig. 17a. It is difficult to extract any simple patterns from the distribution, beyond those discussed above for the folding characteristics. One can, however, use the array as a look-up table to predict the properties of new k-turns, or to relate the sequences to probable function. One point that emerges from inspection of the array is that 3b:3n = A:G is the only cell associated with folding in metal ions and an N3 conformation. If these properties are both required, then that is the only sequence that will confer them. We will take four classes of k-turn and analyze them in terms of their 3b:3n sequence. 4 × 4 arrays of the distributions of 3b:3n sequences may be found in the Supplementary Information of Huang and Lilley (Huang et al. Reference Huang, Wang and Lilley2016). For each group of k-turns we present the distribution of 3b:3n sequences according to their folding and conformational properties in the form of bar charts in Fig. 17b.

Fig. 17. The influence of the 3b:3n sequence on conformation and folding of k-turns. (a) A summary 4 × 4 array where the rows are the 3b and the columns the 3n sequences, combining effect of the identity of the 3b and 3n positions on both N3 or N1 conformation (Huang et al. Reference Huang, Wang and Lilley2016), and whether or not the k-turn will fold in response to addition of metal ions (McPhee et al. Reference Mcphee, Huang and Lilley2014). The latter is shown by color as in Fig. 16, where red cells denote folding in metal ions and blue ones denote failure to fold on the addition of metal ions. The resulting conformation is shown as the collective result from the analysis of natural k-turns and the HmKt-7 variants as shown in Fig. 4. In the case of 3b:3n = A:G, all but one (i.e. HmKt-7 within the ribosome) are N3 structures so this cell is designated N3. The 3b:3n = A:U cell is provisionally assigned as N3 in the light of the distribution of U4snRNA sequences. (b) Bar plots showing the occurrence of 3b:3n sequence variation for four k-turns (Huang et al. Reference Huang, Wang and Lilley2016). For each k-turn type two bar plots are shown. The top one shows the fraction of 3b:3n sequences that confer N3 (blue) vs N1 (green) conformation. The lower one shows the fraction of 3b:3n sequences that confer ion-induced folding (red) v s. inability to fold in metal ions (yellow).
Ribosomal Kt-46. Analysis of 3181 sequences showed that Kt-46 has an extremely strong 3b:3n sequence bias, with only five cells of the 4 × 4 array having non-zero populations. 99·7% are 3b:3n = A:G, giving ion-depended folding, and >99·9% have 3b:3n conferring the N3 conformation. Clearly, Kt-46 is required to fold in ions alone and adopt the N3 conformation. As noted above, 3b:3n = A:G is the only cell that confers both properties and thus Kt-46 is strongly biased in its 3b:3n sequence.
Bacterial ribosomal Kt-7. In contrast to Kt-46, analysis of 2722 Kt-7 sequences showed that >99% have 3b:3n sequences that favor the N1 conformation, of which >92% have 3b:3n = U:G. In addition, 99·7% of the sequences conform to either 3b = C or 3n = G, conferring ion-induced folding. So we deduce that bacterial Kt-7 is required to adopt the N1 conformation and fold in response to ions alone. It is interesting that in the archaeal 50S ribosomal subunit HmKt-7 (which has the N3-conferring 3b:3n = A:G sequence) is forced to adopt an N1 conformation (section 4·1).
U4 snRNA k-turns. Sequence analysis for 9235 U4 snRNA k-turns reveals that all 16 possible 3b:3n are found, although 12 of these are populated to <1·4%. All the red colored cells (i.e. those with 3b:3n sequences that confer ion-induced folding) are avoided, with all but one populated at ⩽0·3%. 63·6% have 3b:3n = G:C, conferring no folding in metal ions and an N3 conformation. The next largest (22·7%) is 3b:3n = G:U, with the same folding and conformational properties. 3b:3n = A:C is populated at 2·4%, so that ⩾88·6% should exhibit weak or non-existent folding in metal ions and adopt the N3 conformation. Interestingly, the third largest fraction of sequences (7·1%) are in the 3b:3n = A:U cell, another non-ion-folding cell but with an unassigned conformation. It, therefore, seems probable that the 3b:3n = A:U sequence also confers the N3 conformation, so this has been included provisionally in Fig. 17a, although this remains unconfirmed by structural analysis at present. If that is true, however, then the fraction of U4 snRNA k-turns with an N3 conformation and weak or zero folding on the addition of metal ions would rise to a total of >95%.Footnote 1
The SAM-I riboswitch k-turn. The SAM-I riboswitch k-turns have the broadest distribution of sequence and properties. Analysis of 4755 sequences showed that 73·4% have 3b:3n sequences that should confer an N3 conformation, while 21·7% should adopt an N1 conformation. We had previously noted that both N3 and N1 conformation k-turns could be accommodated into the SAM-I riboswitch, making it a rather useful experimental vehicle for the structural study of k-turns. ⩾86·6% of the sequences should fold in the presence of metal ions, and indeed 81·7% conformed either to 3b = C or 3n = G. The most populated (60%) cell of the 4 × 4 array is 3b:3n = A:G. Thus the majority of the SAM-I riboswitch k-turns should adopt the N3 conformation and undergo folding in metal ions, but there is a significant minority that is likely to adopt the N1 conformation.
Overall we see that the ribosomal and SAM-I riboswitch k-turns have evolved 3b:3n sequences that are consistent with folding in metal ions alone. For the riboswitches, there are no known binding proteins that might assist folding, so they are probably required to fold unaided. So we might expect that k-turns within riboswitches would generally be required to fold in response to metal ions. In the lysine riboswitch the highest frequency 3b:3n = U:G > A:G > C:C. The majority of the ribosomal k-turns are bound by protein, and yet perhaps during the biogenesis of the ribosome they are required to adopt the correct stable fold before protein binding. We see that both Kt-46 and Kt-7 have 3b:3n sequences that predispose the k-turns to fold in metal ions, despite being of different conformations (N3 and N1 respectively). In the H. marismortui 50S ribosomal subunit Kt-46, Kt-7, Kt-58, Kt-78 and the k-junction J4,5 all have 3b:3n = A:G. The k-turn of the human U4 snRNA contrasts with the ribosomal and riboswitch k-turns in that it has evolved 3b:3n sequences that prevent ion-induced folding. Instead, they have selected 3b:3n sequences that confer N3 conformation but will not fold in metal ions alone. These will be unable to fold stably until the L7Ae or 15·5k protein binds. We conclude that during the formation of the spliceosomal B-complex the folding is incomplete until protein binding has occurred. The sequence of the RNA ensures that it cannot adopt a stable fold in the absence of the protein. Note that there is a second way to achieve a lack of ion-dependent folding so that a k-turn must await the binding of a protein. Some of the box C/D k-turns (e.g. that of A. fulgidus) have selected −1b:−1n = G:C, so fail to fold on the addition of metal ions despite having 3b:3n = U:U (section 10.3).
Another interesting case is that of the archaeal k-turns in the 5′-UTR of the mRNA encoding L7Ae protein (introduced in section 2). The 5′-terminus of the mRNA folds into a hairpin that includes the Shine-Dalgarno sequence that overlaps the bulged strand of a standard simple k-turn. If L7Ae binds to the k-turn it should prevent association with the ribosome, thereby repressing its own translation (Daume et al. Reference Daume, Uhl, Backofen and Randau2017). A corresponding k-turn-containing stem-loop is found in a wide variety of archaea, including Crenarchaeota and Euryarchaeota, and it is instructive to examine the predicted folding properties of these. The repression mechanism would suggest a requirement that the k-turn does not fold until the L7Ae protein binds, i.e. they should not undergo ion-induced folding. The k-turn of Sulfolobus acidocaldarius has 3b:3b = A:U, and −1b:−1n = G:C, both of which confer absence of folding in metal ions alone. The corresponding k-turns of A. fulgidus and Methanosarcina acetivorans also have 3b:3b = A:U, while that of Thermococcus kodakaraensis have 3b:3b = G:U, so all are non-ion-folding k-turns. These have the folding properties anticipated for a genetic switch regulated by the binding of the gene product L7Ae. We also note that all the 3b:3b sequences are predicted to confer an N3 conformation.
11.1 Application of the folding rules to a set of k-turns of unknown structure
As an exercise, we can apply the empirical set of rules to the putative k-turn sequences (Fig. 11) that we identified in the structured RNA species recently presented by Breaker and co-workers as discussed in section 6.5. The predicted properties are summarized in Table 2. Apart from HOLDH the k-turns have −1b:−1n = C:G and three of the four also have A:G as the most frequent 3b:3n sequence. Thus most are expected to fold in response to the addition of metal ions. The k-turn identified in the Actinomyces-1 sequence has 3b:3n = C:C > C:U indicating that this will also fold in response to the presence of metal ions. The folding properties of the HOLDH k-turn are harder to predict because the −1b:−1n = A:U has not been studied experimentally; however it has the 3b:3n = A:G sequence that is the best for ion-induced folding.
The drum and RAGATH-1 k-turns have 3b:3n sequences indicating a strong tendency to adopt the N3 conformation. By contrast, the Actinomyces-1 k-turn has 3b:3n sequences indicating an N1 conformation. HOLDH is the least clear case. The most frequent 3b:3n sequence is A:G, conferring N3 conformation, whereas the next highest is C:A that confers N1 conformation.
The DUF-3268 k-junction has 3b:3n = C:G. If this follows the behavior of the simple k-turns then it should not fold in metal ions and adopt an N3 conformation. However, the A. thaliana TPP riboswitch k-junction also has 3b:3n = C:G yet adopts an N1 conformation (see section 6.3). That indicates the rules cannot be applied to k-junctions with confidence. It seems that the k-junctions have a strong tendency to adopt the N1 conformation, and thus we expect this one to do so as well.
12. Effect of N6-methyladenine inclusion on k-turn folding of snoRNAs
Using X-ray crystallography we have recently shown that trans sugar edge-Hoogsteen G:A basepairs are disrupted if the adenine nucleobase is methylated at N6 (Huang et al. Reference Huang, Ashraf, Wang and Lilley2017). This is in marked contrast to cis-Watson-Crick A:U and A:G basepairs that form normally with N6-methyladenine (Fig. 18). Since trans sugar edge-Hoogsteen G:A basepairs form the core of the k-turn it is perhaps unsurprising that N6-methylation of the 1n adenine prevents folding of HmKt-7 (S. Ashraf, LH and DMJL, unpublished data). In principle, this might form the basis of a regulatory mechanism controlled by reversible methylation in cells.

Fig. 18. The effect of N6-methyladenine (N6mA) inclusion on trans sugar-Hoogsteen G:A base pairing, and its potential effect on box C/D snoRNP assembly (Huang et al. Reference Huang, Ashraf, Wang and Lilley2017). (a) and (b) Crystal structures of G:A (PDB ID 5LR3; at 1·65 Å resolution) and G: N6mA (PDB ID 5LR4 at 1·72 Å resolution) base pairs in a duplex as tandem G:A, A:G pairs flanked by G:U base pairs. The unmodified G:A base pairs (a) form sheared trans-sugar-Hoogsteen G:A base pairs like those found in the k-turns. By contrast the in the G:6mA pair (b) there are no H-bonds connecting the nucleobases but rather N6mAN6 forms an H-bond with GO2′. (c) Binding of human 15·5k protein (L7Ae ortholog) to the box C/D k-turn of human U13 snRNA. The sequence of the k-turn is shown. The RNA was prepared with either adenine or N6mA at the 1n position of the k-turn. Radioactive RNA was incubated with increasing concentrations of 15·5k protein, and free RNA and complexes with bound protein separated by electrophoresis in polyacrylamide. 15·5k-bound complexes migrate as retarded species. RNA with unmodified adenine (tracks 1–4) binds 15·5k protein as a discrete retarded complex, with a smear of non-specific binding at the highest protein concentration (track 4). By contrast, RNA with N6mA at the 1n position (tracks 5–8) does not give rise to a specific complex, but only non-specific binding. (d) Analysis of 15·5k protein-induced folding of human U13 snRNA using steady-state FRET. FRET efficiency (E FRET) was measured using RNA terminally labeled with fluorescein donor and Cy-3 acceptor analogous to that in Fig. 12. The RNA was prepared with either adenine (filled circles) or N6mA (open circles) at the 1n position of the k-turn. E FRET was measured as a function of added 15·5k protein concentration, and the data fitted to a simple binding isotherm. That for the unmodified RNA undergoes a two-state folding process similar to that shown for AfL7Ae binding to HmKt-7 (Fig. 12b). In contrast, the N6mA-containing RNA requires a higher concentration of 15·5k protein and achieves a significantly lower end-point. It is likely that this corresponds to the non-specific binding observed in the electrophoretic experiments shown in part b.
The principal methyl transferase in eukaryotes that is responsible for generating N6-methyladenine in RNA is METTL3-METTL14 (Liu et al. Reference Liu, Yue, Han, Wang, Fu, Zhang, Jia, Yu, Lu, Deng, Dai, Chen and He2014). This has the preferred target DRACH (where D denotes A, G or U, R is A or G and H is A, C or U), with GAC as the most common site of methylation (Dominissini et al. Reference Dominissini, Moshitch-Moshkovitz, Schwartz, Salmon-Divon, Ungar, Osenberg, Cesarkas, Jacob-Hirsch, Amariglio, Kupiec, Sorek and Rechavi2012; Meyer et al. Reference Meyer, Saletore, Zumbo, Elemento, Mason and Jaffrey2012a). The central A is the site of methylation. Thus for the A1n of a k-turn to become methylated would require −1n to be cytosine, yet in the majority of k-turns −1b:−1n = C:G. However, we had noted that in some box C/D snoRNA k-turns the −1b:−1n base pair is inverted, so that −1n = C thus creating the required GAC methylation target. We recently performed a bioinformatic data mining analysis of human box C/D and C′/D′ snoRNA sequences, finding that 27 have the required GAC target sequence, of which 14 have been found to be methylated in the cell (Huang et al. Reference Huang, Ashraf, Wang and Lilley2017). Thus a significant sub-set of human box C/D and C′/D′ k-turn sequences undergo adenine N6-methylation at the critical A1n position.
Assembly of the box C/D snoRNP species requires the ordered association of a series of proteins with the RNA (Bleichert et al. Reference Bleichert, Gagnon, Brown, Maxwell, Leschziner, Unger and Baserga2009; Kiss-Laszlo et al. Reference Kiss-Laszlo, Henry, Bachellerie, Caizergues-Ferrer and Kiss1996; Lin et al. Reference Lin, Lai, Jia, Xu, Zhang, Lu and Ye2011; Tran et al. Reference Tran, Zhang and Maxwell2003; Tycowski et al. Reference Tycowski, Smith, Shu and Steitz1996; Watkins et al. Reference Watkins, Segault, Charpentier, Nottrott, Fabrizio, Bachi, Wilm, Rosbash, Branlant and Luhrmann2000; Xue et al. Reference Xue, Wang, Yang, Terns, Terns, Zhang, Maxwell and Li2010; Ye et al. Reference Ye, Jia, Lin, Ju, Peng, Xu and Zhang2009). In the first stage, the 15·5k protein binds to the k-turns. Once this has occurred the next proteins (Nop56 and Nop58 in eukaryotes) bind. These are analogous to Prp31 that binds to the complex of 15·5k with the U4 k-turn in the spliceosomal B complex (Liu et al. Reference Liu, Li, Dybkov, Nottrott, Hartmuth, Luhrmann, Carlomagno and Wahl2007) (see section 2). Lastly, two molecules of the 2′O-methyl transferase fibrillarin associate with the complex to generate the catalytically-active snoRNP. Importantly Watkins et al. (Reference Watkins, Dickmanns and Luhrmann2002) found that if the binding of 15·5k to the human box C/D k-turn is prevented by sequence changes known to interfere with k-turn folding, assembly of the box C/D snoRNP does not occur. We showed in vitro that inclusion of N6-methyladenine at the 1n position of some representative human box C/D k-turns both prevented specific 15·5k protein binding, and the proper folding of the k-turn (Huang et al. Reference Huang, Ashraf, Wang and Lilley2017) (Fig. 18). While this is perhaps a step away from demonstrating an in vivo regulatory mechanism based on reversible methylation of k-turn RNA, it is very plausible and further experiments to test this are under way.
13. The k-turn as a building unit in nano-construction
The precise geometry of the k-turn suggests a possible use as a building block in the construction of molecular nano-objects (Jasinski et al. Reference Jasinski, Haque, Binzel and Guo2017). RNA has previously been used as a material for nano-scale engineering, and helical bulges have been used to construct a square or triangular molecules (Boerneke et al. Reference Boerneke, Dibrov and Hermann2016; Dibrov et al. Reference Dibrov, Mclean, Parsons and Hermann2011; Ohno & Inoue, Reference Ohno and Inoue2015). Moreover, this provides a test of our conformational understanding, whether or not we can manipulate the structure at will to design and generate molecular assemblies. Saito and colleagues (Ohno et al. Reference Ohno, Kobayashi, Kabata, Endo, Iwasa, Yoshimura, Takeyasu, Inoue and Saito2011) made the first steps on this path, using atomic force microscopy (AFM) to visualize triangular objects comprising three k-turns bound by the L7Ae protein.
13.1 Association of a two-k-turn unit in crystal lattices
The basic structural element used in our studies comprised a two-fold symmetric duplex with two HmKt-7 sequences (Fig. 19a) (Huang & Lilley, Reference Huang and Lilley2016). Their loops are located on opposite strands and the k-turns are associated through their NC helices, with ten basepairs separating the two loops, including the G:A pairs. The structure had been determined by X-ray crystallography at high resolution, revealing it to have an overall horse-shoe shape (McPhee et al. Reference Mcphee, Huang and Lilley2014). It has a central dyad but is not fully planar. Within the structure, the HmKt-7 elements adopt their standard N3 k-turn structure. The potential of this two-k-turn unit (the 2 K unit) became apparent when intermolecular contacts between units within the crystal lattice were examined. In fact, two crystal forms were obtained. One had tetragonal symmetry (P4222), where two 2 K units form a dumbbell shape. The second had hexagonal symmetry (P6322), and in this case, three 2 K units were associated to form a triangular shape (Fig. 19a). In both cases, the interaction between molecules occurred by end-to-end coaxial stacking of the C-helices, with preservation of helical symmetry and stacking (base plane separation of 3·3 Å).

Fig. 19. The k-turn as a unit for nano-construction. The structure of a triangular molecular object comprising six k-turns (Huang & Lilley, Reference Huang and Lilley2016). (a) The two k-turn unit (2 K unit) is a duplex with two HmKt-7 sequences related by a two-fold rotation so the loops are on opposite strands and they are connected by their common NC helix (PDB ID 4CS1; at 2·0 Å resolution). They pack in two ways to form a crystal lattice. The parallel-eye stereoscopic image shows that in which three 2 K units associated by end-to-end stacking, related by the three-fold rotation axis shown. (b) A single RNA duplex containing three 2 K units, i.e. six HmKt-7 sequences with alternating polarity. In the crystal, the molecule adopts a triangular conformation with a random rotational setting within the lattice creating a crystallographic three-fold rotation axis as shown (PDB ID 5G4 T; at 2·75 Å resolution). Thus the molecule has a pseudo-symmetry with symmetry point group D 3.
Two further structures were obtained for 2 K units comprising HmKt-7 with altered 3b:3n sequences, crystallized as complexes with bound AfL7Ae protein. That with 3bU:3nU crystallized with orthorhombic symmetry (P212121), and formed a triangular association in the lattice based on coaxial end-to-end stacking as before. However, the central pore was two-fold larger than that found for the protein-free triangular structure based on unmodified Kt-7. By contrast, the 2 K unit based on HmKt-7 3bG:3nC crystallized with monoclinic symmetry (C1 2 1), and in this case, four 2 K units (i.e. 8 k-turns and 8 L7Ae molecules) adopted a square assembly by coaxial end-to-end stacking. Thus by small changes of the sequence the geometry and dimensions of the assemblies can be radically altered.
13.2 The molecular structure of a six-k-turn nano-object
With this background and the experience of manipulating the structural properties of the basic building block a single duplex RNA was synthesized containing three consecutive 2 K using (derived from a single strand of 57 nt), i.e. six HmKt-7 motifs alternating in polarity. This crystallized without requiring the addition of L7Ae, in a hexagonal space group (P6322), diffracting to 2·75 Å resolution (Huang & Lilley, Reference Huang and Lilley2016). The molecule folded into a triangular shape with end-on-end coaxial stacking at the open end (Fig. 19b). Remarkably, this was so perfectly formed that the individual molecules crystallized with static disorder, making no distinction between a covalently continuous junction between C helices and the open end. Thus each phosphate group connecting the two-k-turn units had an occupancy of 2/3. Effectively the molecule has an apparent point group symmetry D 3, with a shape reminiscent of a cyclohexane molecule in its chair conformation.
These studies demonstrate that the k-turn is an effective and versatile building block in the assembly of nano-scale molecular objects, and demonstrates just why k-turns are widely used in natural RNA molecules to mediate tertiary contacts and organize architecture over a large scale. The k-turn is one of nature's most useful nano-scale structure-building elements in functional RNA molecules.
14. In conclusion
The k-turn is an extremely widespread structural motif in RNA architecture, involved in the construction of many key RNA structures of the cell that are important in translation, RNA processing, and genetic control. We have seen that the formation of k-turns is the essential first step in the formation of RNA-protein assemblies such as the box C/D snoRNP and that this can be controlled by methylation of a key adenine nucleotide in the k-turn. The k-turn structure has been the subject of intense study for the last decade or more so that it is arguably the most well-understood RNA motif. We now comprehend these structures in considerable detail, and how folding and conformational properties are determined by local sequence. We see how the sequences of individual k-turns fit their structural properties to the biological requirements of their situation. Moreover, we now understand the structure to the point at which we can use it as the basis for the design of defined nano-structural objects. This perhaps is no surprise, since the k-turn is essentially a major component of nature's nano-technological construction set.
Acknowledgements
We thank Terry Goody, Jia Liu, Ben Turner, Tim Wilson, Peter Daldrop, Kersten Schroeder, Jia Wang, Jo Ouellet, Scott McPhee and Saira Ashraf for their contributions to studies of k-turn structures in RNA, and all past and present members of the DMJL lab for many discussions. We thank our collaborators, especially Xuesong Shi, Dan Herschlag and Benoit Masquida, and Cancer Research UK, the Wellcome Trust and the HFLP for funding. X-ray crystallographic data have been collected at the Diamond and ESRF synchrotrons.