
Contents
Research Article
The ICDD/PDF Coverage of the High Tc Superconductor and Related Compounds in the A-R-Cu-O Systems (A = Ba, Sr and Ca, and R = Lanthanides and Y)
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 125-133
-
- Article
- Export citation
The Use of Reference Intensity Ratios in X-Ray Quantitative Analysis
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 186-193
-
- Article
- Export citation
A Study of Grazing Incidence Configurations and Their Effect on X-Ray Diffraction Data
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-5
-
- Article
- Export citation
The Background in X-Ray Powder Diffractograms: A Case Study of Rietveld Analysis of Minor Phases Using Ni-Filtered and Graphite-Monochromated Radiation
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 63-70
-
- Article
- Export citation
XPAS: An Interactive Computer Program for Analysis of X-Ray Powder Diffraction Patterns
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 6-10
-
- Article
- Export citation
The Determination of Low Levels of Quartz in Commercial Kaolins by X-ray Diffraction
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 71-76
-
- Article
- Export citation
On Indexing X-Ray Diffraction Powder Patterns of Cubic Lazurites with an Incommensurate-Modulated Structure
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 134-136
-
- Article
- Export citation
Simultaneous Determination of Layer Thickness, Composition, and Mass Absorption by X-Ray Diffraction
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 194-196
-
- Article
- Export citation
Intermediate Phases Produced During the Formation of Cadmium-Nickel Ferrites
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 11-12
-
- Article
- Export citation
About Instrument-, Method- and Matrix-Independence in the Working Equations of the Quantitative X-Ray Analysis
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 197-199
-
- Article
- Export citation
A Simplified Correction for Surface Roughness Effects in X-Ray Diffractometric Investigations
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 77-82
-
- Article
- Export citation
Interlaboratory Tests of X-Ray Quantitative Phase Analysis
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 137-141
-
- Article
- Export citation
X-Ray Determination of a Crystal System
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 200-205
-
- Article
- Export citation
A New Method Of X-Ray Crystal Analysis*
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 13-27
-
- Article
- Export citation
A Review of the XRD Data of the Phases Present in the CaO-SrO-CuO System
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 142-148
-
- Article
- Export citation
Notes on Slits and Monochromators in Accurate Powder Diffractometry
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 83-88
-
- Article
- Export citation
Structure Determination of Tl4V2O7 from Powder Diffraction Data using an Inel X-Ray PSD: Stereochemical Activity of Thallium(I) Lone Pair
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 206-211
-
- Article
- Export citation
-
The crystal structure of Tl4V2O7 is solved ab-initio from powder diffraction data collected in Debye-Scherrer geometry using an Inel X-ray Position Sensitive Detector. The structure has been determined from Rietveld analysis in space group
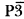 ml, Z = 1, with a = 5.9388(2)Å and c = 7.7322(3)Å. The structure of Tl4V2O7 is built up from isolated V2O7 groups aligned along the trigonal c axis. Thallium atoms alternate along a 3-fold axis. The presence of stereochemically active lone pairs is demonstrated and their positions are calculated using a self-consistent electrostatic model. The influence of sample absorption is briefly discussed and the results are compared with those obtained in Bragg-Brentano geometry using flat-plate specimen.
ml, Z = 1, with a = 5.9388(2)Å and c = 7.7322(3)Å. The structure of Tl4V2O7 is built up from isolated V2O7 groups aligned along the trigonal c axis. Thallium atoms alternate along a 3-fold axis. The presence of stereochemically active lone pairs is demonstrated and their positions are calculated using a self-consistent electrostatic model. The influence of sample absorption is briefly discussed and the results are compared with those obtained in Bragg-Brentano geometry using flat-plate specimen.
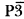 ml, Z = 1, with a = 5.9388(2)Å and c = 7.7322(3)Å. The structure of Tl
ml, Z = 1, with a = 5.9388(2)Å and c = 7.7322(3)Å. The structure of TlX-Ray Powder Diffraction Data for (1R,2R) -(−)-norpseudoephedrine hydrochloride, (lS,2R)-(+)-norephedrine hydrochloride, and (±)-norephedrine hydrochloride
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 28-31
-
- Article
- Export citation
A Geometric Factor for Asymmetric Diffraction
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 149-151
-
- Article
- Export citation
Intensity Calibration Curves for Bragg-Brentano X-Ray Diffractometers
-
- Published online by Cambridge University Press:
- 10 January 2013, pp. 89-94
-
- Article
- Export citation