



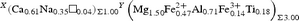
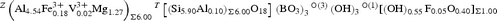
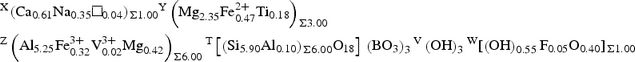
Introduction
Tourmalines are complex borosilicates that have been studied extensively in terms of their crystal structure and crystal chemistry (e.g. Foit, Reference Foit1989; Hawthorne, Reference Hawthorne1996; Hawthorne and Henry, Reference Hawthorne and Henry1999; Ertl et al., Reference Ertl, Hughes, Pertlik, Foit, Wright, Brandstatter and Marler2002; Novák et al., Reference Novák, Povondra and Selway2004; Bosi, Reference Bosi2013, Reference Bosi2018; Henry and Dutrow, Reference Henry and Dutrow2011; Cempírek et al., Reference Cempírek, Houzar, Novák, Groat, Selway and Šrein2013; Bačík and Fridrichová, Reference Bačík and Fridrichová2021). In accordance with Henry et al. (Reference Henry, Novák, Hawthorne, Ertl, Dutrow, Uher and Pezzotta2011), the general chemical formula of tourmaline is written as: XY3Z6T6O18(BO3)3V3W, where X = Na+, K+, Ca2+, □ (= vacancy); Y = Al3+, Fe3+, Cr3+, V3+, Mg2+, Fe2+, Mn2+, Li+; Z = Al3+, Fe3+, Cr3+, V3+, Mg2+, Fe2+; T = Si4+, Al3+, B3+; B = B3+; V = (OH)–, O2–; and W = (OH)–, F–, O2–. Note that the letters X, Y, T, Z and B represent groups of cations at the [9]X, [6]Y, [6]Z, [4]T and [3]B crystallographic sites (designated by italicised letters). The letters V and W in the formula represent groups of anions accommodated at the [3]-coordinated O(3) and O(1) crystallographic sites, respectively. The dominance of specific ions at one or more sites of the structure gives rise to numerous distinct mineral species.
A formal description of the new tourmaline species uvite is presented here. Uvite is a common mineral name in the tourmaline literature and refers to the province of Uva (Sri Lanka) as the type locality for the formerly supposed occurrence of this mineral (Kunitz, Reference Kunitz1929). However, the tourmaline from the Uva locality should actually correspond to a fluor-species (Dunn et al., Reference Dunn, Appleman, Nelen and Norberg1977). In accord with the tourmaline nomenclature scheme, the root name of fluor-uvite (Henry et al., Reference Henry, Novák, Hawthorne, Ertl, Dutrow, Uher and Pezzotta2011) requires that the name uvite should be given to the hydroxy analogue. The approval of this new species has been beset by difficulties. Uvite was approved by the International Mineralogical Association's Commission on New Minerals, Nomenclature and Classification (IMA–CNMNC) as a valid mineral species with the proposal no. 2000-030a (Clark et al., Reference Clark, Hawthorne and Grice2010), but the complete description has never been published. This proposal was subsequently withdrawn as additional analytical work done by the authors of proposal 2000-30a showed this material to be a potentially new oxy-tourmaline (Hålenius et al., Reference Hålenius, Hatert, Pasero and Mills2018). In July 2019, another proposal (no. 2019-004) was rejected. Finally, uvite has been approved by the IMA–CNMNC with proposal no. 2019-113 (Bosi et al., Reference Bosi, Biagioni, Pezzotta, Skobgy, Hålenius, Cempírek, Hawthorne, Lussier, Abdu, Day, Fayek, Clark, Grice and Henry2020) using a specimen collected at Facciatoia, at the eastern limit of the village of San Piero in Campo, Elba Island, in an abandoned magnesite quarry in 2019 by the mineral collector Michele Degl'Innocenti. Holotype material is deposited in the collections of the Natural History Museum of Milano, Italy, catalogue number M38848, and the Museo di Storia Naturale, University of Pisa, catalogue number 19911.
Occurrence
The holotype specimen was collected from a narrow vein of aggregates of dark brown-to-black tourmaline, penetrating hydrothermally altered metaserpentinite rich in magnesite and dolomite, in the abandoned Facciatoia quarry, east of the village of San Piero in Campo, Elba Island, Livorno, Tuscany, Italy (42°45′04.55″N, 10°12′50.89″E). Facciatoia is a classic mineralogical locality, in which several narrow LCT pegmatites rich in multi-coloured and pink elbaite crystals were found in the past (today the locality is exhausted; Pezzotta, Reference Pezzotta2021); the pegmatites crosscut a lens of porphyritic monzogranite and the surrounding hydrothermally altered metaserpentinites.
In San Piero in Campo, tourmaline-rich veins typically cross-cut metaserpentinites around miarolitic tourmaline-bearing pegmatites in the metamorphic aureole of the Monte Capanne intrusion. These veins are up to 2 or 3 cm wide and can be up to a few metres long. They are entirely composed of tourmaline-supergroup minerals (uvite and magnesio-lucchesiite), and locally form small cavities in which tourmaline occurs as blackish sharp and lustrous short prisms, up to 1 cm long and 5 mm in diameter.
Appearance, physical and optical properties
Uvite occurs as massive vein-filling subhedral grains and rare euhedral crystals up to 1 cm in size and is brown with a vitreous lustre (Fig. 1). The morphology consists of {10$\bar{1}$ 0} and {11$\bar{2}$
0} and {11$\bar{2}$ 0} prisms terminated by {10$\bar{1}$
0} prisms terminated by {10$\bar{1}$ 1} and {10$\bar{1}\bar{1}$
1} and {10$\bar{1}\bar{1}$ } pyramidal faces. Prism faces are striated parallel to the c axis. It has a grey streak and shows no fluorescence, has a Mohs hardness of ~7½ (estimated by analogy with magnesio-lucchesiite; Scribner et al., Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) and is brittle with a conchoidal fracture. The calculated density, based on the empirical formula and unit-cell volume refined from single-crystal X-ray diffraction (XRD) data, is 3.115 g/cm3. In thin section, uvite is transparent; in transmitted light, it is pleochroic, O = greenish brown, E = pale yellow, with O > E. Uvite is uniaxial (–) with refractive indices ω = 1.660(5) and ɛ = 1.640(5) measured by the immersion method using white light from a tungsten source. The mean index of refraction, density, and chemical composition resulted in an excellent compatibility index (1 – Kp/Kc = 0.021) (Mandarino, Reference Mandarino1981).
} pyramidal faces. Prism faces are striated parallel to the c axis. It has a grey streak and shows no fluorescence, has a Mohs hardness of ~7½ (estimated by analogy with magnesio-lucchesiite; Scribner et al., Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) and is brittle with a conchoidal fracture. The calculated density, based on the empirical formula and unit-cell volume refined from single-crystal X-ray diffraction (XRD) data, is 3.115 g/cm3. In thin section, uvite is transparent; in transmitted light, it is pleochroic, O = greenish brown, E = pale yellow, with O > E. Uvite is uniaxial (–) with refractive indices ω = 1.660(5) and ɛ = 1.640(5) measured by the immersion method using white light from a tungsten source. The mean index of refraction, density, and chemical composition resulted in an excellent compatibility index (1 – Kp/Kc = 0.021) (Mandarino, Reference Mandarino1981).

Fig. 1. Photo of the holotype fragment (#19911) of uvite in transmitted light.
Analytical methods and results
Microprobe analysis
Electron microprobe analysis was done using a wavelength-dispersive spectrometer (WDS mode) with a Cameca SX50 instrument at the ‘Istituto di Geologia Ambientale e Geoingegneria, CNR (Rome, Italy)’, operating at an accelerating potential of 15 kV, a sample current of 15 nA and 10 μm beam diameter. The following standards, X-ray Kα lines and analyser crystals were used: wollastonite (Si and Ca; PET), magnetite (Fe; LIF), rutile (Ti; PET), corundum (Al; TAP), vanadinite (V; PET), fluorophlogopite (F; TAP), periclase (Mg; TAP), jadeite (Na; TAP), orthoclase (K; PET), sphalerite (Zn; LIF), chromium oxide (Cr; PET), and metallic Mn (Mn; LIF). The PAP routine was applied (Pouchou and Pichoir, Reference Pouchou, Pichoir, Heinrich and Newbury1991). Table 1 gives mean values of 15 spot analyses. Chromium, Mn, Zn and K were below detection limits (<0.03 wt.%).
Table 1. Electron microprobe data (WDS mode) and atoms per formula unit (apfu) normalised to 31 anions for uvite.

a Calculated by stoichiometry, (Y + Z+T) = 15.00 apfu.
b Determined by Mössbauer spectroscopy
S.D. – standard deviation
Mössbauer spectroscopy
Mössbauer spectroscopy (MS) was used to determine the Fe3+/ΣFe ratio of the sample, using a conventional spectrometer system operated in constant acceleration mode (Swedish Museum of Natural History, Stockholm, Sweden). The absorber was prepared from 40 mg of ground sample mixed with an acrylic resin which was pressed to a 12-mm diameter disc under mild heating (<150°C). Data were collected over 1024 channels in the velocity range –4.2 to +4.2 mm/s using a 57Co rhodium matrix standard source of 50 mCi nominal activity and were calibrated and folded against the spectrum of an α-Fe foil. The spectrum (Fig. 2) was fit using the software MossA (Prescher et al., Reference Prescher, McCammon and Dubrowinsky2012) with three doublets assigned to Fe2+ and one doublet assigned to Fe3+, resulting in an Fe3+/ΣFe ratio of 0.40, assuming similar recoil-free fractions for Fe2+ and Fe3+ (Table 2).

Fig. 2. Mössbauer spectrum of uvite. Fitted absorption doublets assigned to Fe2+ and Fe3+ are indicated in blue and red, respectively. Diamonds denote the measured spectrum, and the black curve represents summed fitted spectra.
Table 2. Mössbauer parameters for uvite obtained at room-temperature.*

* δ = centroid shift, ΔEQ = quadrupole splitting, FWHM = full width at half-maximum, A = relative area.
Single-crystal infrared spectroscopy
Polarised Fourier-transform infrared (FTIR) absorption spectra were measured on a 39 μm thick doubly polished single-crystal section oriented parallel to the c-axis. A Bruker Vertex 70 spectrometer attached to a Hyperion 2000 microscope was used to collect spectra in the range 2000–13000 cm–1 at a resolution of 4 cm–1 (Swedish Museum of Natural History, Stockholm, Sweden). Spectra recorded in polarised mode parallel to the crystallographic c-axis show a very intense band at ~3570 cm–1, which is off-scale due to excessive absorption, and three less-intense bands at 3666, 3728 and 3764 cm–1 (Fig. 3). A shoulder occurs around 3623 cm–1. Spectra obtained perpendicular to the c-axis show considerably weaker bands at 3577 and 3623 cm–1, which may be the bands responsible for the very strong absorption in the opposite polarisation direction.

Fig. 3. Polarised FTIR spectra for uvite. Note the presence of bands above 3650 cm–1. The main band is truncated at ~2 absorbance units in the E‖c direction due to excessive absorption. Sample thickness 39 μm.
Bands above about 3600–3650 cm–1 are usually due to (OH) at the O(1) site (≡ W) (e.g. Gonzalez-Carreño et al., Reference Gonzales-Carreño, Fernández and Sanz1988; Bosi et al., Reference Bosi, Skogby, Lazor and Reznitskii2015a). Based on the empirical crystal-chemical formula (see below) and the studies of Watenphul et al. (Reference Watenphul, Burgdorf, Schlüter, Horn, Malcherek and Mihailova2016) and Bosi et al. (Reference Bosi, Skogby, Hålenius and Ciriotti2018), the main FTIR bands at ~3577 cm–1 are probably caused by the atomic arrangements {2[(Fe,Mg)AlAl]–(AlAlAl)}–O(3)(OH)3, whereas we prefer to ascribe the band at ~3623 to the arrangements 3[(Fe,Mg)AlAl]–O(3)(OH)3 rather than to Y[(Fe,Mg)AlAl]–O(1)(OH)–X(□) because the X□ content is too low. The latter arrangement is more likely to be related to the band at ~3666 cm–1 in the atomic arrangement Y(MgMgAl)–O(1)(OH)–X(□). In this regard, note that the bands between ~3650–3700 cm–1 are associated with X□, whereas those above 3700 cm–1 are associated with X(Na,Ca) (e.g. Gonzalez-Carreño et al., Reference Gonzales-Carreño, Fernández and Sanz1988; Berryman et al., Reference Berryman, Wunder, Ertl, Koch-Müller, Rhede, Scheidl, Giester and Heinrich2016; Watenphul et al., Reference Watenphul, Burgdorf, Schlüter, Horn, Malcherek and Mihailova2016). Consequently, bands at ~3728 and ~3764 cm–1 may be due to the arrangements Y(FeFeAl)–O(1)(OH)–X(Na) and Y(MgMgMg)–O(1)(OH)–X(Na,Ca), respectively.
Optical absorption spectroscopy (OAS)
Polarised optical absorption spectra of uvite (Fig. 4) were acquired at the Natural History Museum of Stockholm, Sweden. Data were collected at room temperature on the same polished crystal that was used for FTIR measurements. An AVASPEC-ULS2048X16 spectrometer, connected via a 400 μm UV fibre cable to a Zeiss Axiotron UV-microscope, was used. A 75 W Xenon arc lamp was used as a light source and Zeiss Ultrafluar 10× lenses served as objective and condenser. A UV-quality Glan-Thompson prism, with a working range from 40000 to 3704 cm–1, was used as the polariser.

Fig. 4. Polarised optical absorption spectra of uvite in the UV and visible region. Sample thickness 39 μm.
The recorded spectra show broad and strongly polarised (O > E) absorption bands at 22000, 14250 and 8790 cm–1 (Fig. 4). In agreement with previous optical studies of tourmaline (e.g. Mattson and Rossman, Reference Mattson and Rossman1987), the bands at 14250 and 8790 cm–1 are assigned to Fe3+-enhanced spin-allowed d–d transitions in [6]-coordinated Fe2+. The broad, intense, and strongly E||O-polarised band at 22000 cm–1 is due to Fe2+-Ti4+ intervalence charge transfer processes (e.g. Smith, Reference Smith1978; Taran et al., Reference Taran, Lebedev and Platonov1993).
Single-crystal structure refinement
A representative crystal of uvite from Facciatoia was selected for XRD measurements on a Bruker APEX-II single-crystal diffractometer, equipped with a Photon II CCD area detector and a graphite-crystal monochromator, using MoKα radiation from a fine-focus sealed X-ray tube (Dipartimento di Scienze della Terra, University of Pisa). The sample-to-detector distance was 5 cm. A total of 1675 exposures (step = 0.2°, time/step = 20 s) covering a full reciprocal sphere with a redundancy of ~15 was collected using ω and φ scan modes. Final unit-cell parameters were refined using the Bruker AXS SAINT program on reflections with I > 10 σI in the range 5° < 2θ < 72°. The intensity data were processed and corrected for Lorentz, polarisation and background effects using the APEX3 software program of Bruker AXS. The data were corrected for absorption using a multi-scan method (SADABS). The absorption correction led to an improvement in R int. No violation of R3m symmetry was detected.
Structure refinement was done using the SHELXL-2013 program (Sheldrick, Reference Sheldrick2015). Starting coordinates were taken from Bosi et al. (Reference Bosi, Skogby, Ciriotti, Gadas, Novák, Cempírek, Všianský and Filip2017a). Variable parameters were as follows: scale factor, extinction coefficient, atom coordinates, site-scattering values (for X, Y and Z sites) and atomic-displacement factors. Attempts to refine the extinction coefficient yielded values within its standard uncertainty, thus it was not refined. Neutral atom scattering factors were used. In detail, the X site was first modelled using Ca versus Na, but this yielded a strong correlation (r = 0.82) between XU 11 and the X site-scattering value; thus, to avoid this correlation, the X site was modelled by setting the Na content to 0.354 atoms per formula unit (apfu, see below) and allowing the remainder of the site to refine as Ca. The occupancy of the Y site was obtained considering the presence of Mg versus Fe, and the Z site with Al versus Fe. The T, B and anion sites were modelled, respectively, with Si, B and O scattering factors and with a fixed occupancy of 1, because refinement with unconstrained occupancies showed no significant deviations from this value. The position of the H(1) and H(3) atoms bonded to oxygen at the O(1) and O(3) sites, respectively, in the structure was taken from the difference-Fourier map and incorporated into the refinement model. In accord with Gatta et al. (Reference Gatta, Bosi, McIntyre and Skogby2014), the O(1)–H(1) and O(3)–H(3) bond lengths were restrained (by DFIX command) to be 0.96 Å and 0.97 Å (respectively) with their isotropic-displacement parameters constrained to be equal to 1.2 times that obtained for the O(1) and O(3) sites. A final refinement was then done by modelling the site occupancy of the O(1) site with O and F fixed at the value obtained from the empirical formula (see below). Similar chemical constraints were applied to refine the H(1) and H(3) sites. There were no correlations greater than 0.7 between the parameters at the end of the refinement. Table 3 lists crystal data, data-collection information, and refinement details; Table 4 gives the fractional atom coordinates, equivalent isotropic-displacement parameters and Table 5 shows selected bond lengths. The crystallographic information file has been deposited with the Principal Editor of Mineralogical Magazine and is available as Supplementary material (see below).
Table 3. Single-crystal X-ray diffraction data details for uvite.*

* Notes: R int = merging residual value; R 1 = discrepancy index, calculated from F-data; wR 2 = weighted discrepancy index, calculated from F 2-data;. Refined as an inversion twin.
Table 4. Fractional atom coordinates, isotropic (*) or equivalent-isotropic displacement parameters (in Å2) and site occupancies for uvite.

* Isotropic-displacement parameters (U iso) for H(1) and H(3) constrained to have a U iso 1.2 times the U eq value of the O(1) and O(3) oxygen atoms, respectively.
Table 5. Selected bond lengths (Å) for uvite.

Powder X-ray diffraction
A powder X-ray diffraction pattern for uvite was collected at the Natural History Museum of Stockholm (Sweden) using a Panalytical X'pert powder diffractometer equipped with an X'celerator silicon-strip detector. The range 5–80° (2θ) was scanned with a step-size of 0.017° with the sample mounted on a background-free Si holder using sample spinning. The diffraction data (for CuKα = 1.54059 Å), corrected using Si as an internal standard, are listed in Table 6. The program UnitCell (Holland and Redfern, Reference Holland and Redfern1997) was used to refine unit-cell parameters in the trigonal system: a = 15.9729(3) Å, c = 7.2291(2) Å and V = 1597.3(1) Å3.
Table 6. Powder X-ray diffraction data for uvite.*

* Only the reflections with I ≥ 5% are listed. The six strongest reflections are given in bold.
Determination of the number of atoms per formula unit
In agreement with the structure-refinement results, the boron content was assumed to be stoichiometric (B3+ = 3.00 apfu). Both the site-scattering results and the bond lengths of B and T are consistent with the B site fully occupied by boron and no amount of B3+ at the T site (e.g. Bosi and Lucchesi, Reference Bosi and Lucchesi2007). The iron oxidation state was determined by MS. In accordance with Pesquera et al. (Reference Pesquera, Gil-Crespo, Torres-Ruiz, Torres-Ruiz and Roda-Robles2016), the Li content was assumed to be insignificant as MgO > 2 wt.% in the sample studied. The (OH) content and the formula were then calculated by charge balance with the assumption (T + Y + Z) = 15 apfu and 31 anions pfu (Table 1). The excellent agreement between the number of electrons per formula unit (epfu) derived from EMPA and SREF (241.2 and 241.1 epfu, respectively) supports the stoichiometric assumptions.
Site populations
The uvite site populations at the X, B, T, O(3) (≡ V) and O(1) (≡ W) sites follow the standard site preference suggested for tourmaline (e.g. Henry et al., Reference Henry, Novák, Hawthorne, Ertl, Dutrow, Uher and Pezzotta2011) and are in accord with the information from FTIR absorption spectra (Fig. 3). The site populations at the octahedrally coordinated Y and Z sites were optimised according to the procedure of Bosi et al. (Reference Bosi, Reznitskii, Hålenius and Skogby2017b), and by fixing the minor elements Ti4+ at Y and V3+ at Z.
The resulting empirical crystal-chemical formula is:

The refined site-scattering values (Hawthorne et al., Reference Hawthorne, Ungaretti and Oberti1995) and those calculated from the site populations are compared in Table 7. The agreement between the refined and calculated values is very good, and validates the distribution of cations over the X, Y and Z sites. These site populations are also supported by the close accord of the weighted bond-valence sums and weighted atom valences (or mean formal charges) calculated from the empirical crystal-chemical formula (Table 8).
Table 7. Refined site-scattering values and optimised site-populations for uvite.

a = electrons per formula unit; b = atoms per formula unit
Table 8. Weighted bond-valences (valence units) for uvite.*

* Notes: Weighted bond valence according to Bosi (Reference Bosi2014); bond valence parameters from Gagné and Hawthorne (Reference Gagné and Hawthorne2015).
a Mean Formal Charge (or weighted atom valence) from the empirical crystal-chemical formula.
For classification purposes, the optimised formula was recast in its ordered form, i.e. with trivalent cations ordered in the Z position of the tourmaline general chemical formula (Henry et al., Reference Henry, Novák, Hawthorne, Ertl, Dutrow, Uher and Pezzotta2011):
X(Ca0.61Na0.35□0.04)Σ1.00 Y(Mg2.35Fe2+ 0.47Ti0.18)Σ3.00 Z(Al5.25Fe3+ 0.32V3+ 0.02Mg0.42)Σ6.00 T[(Si5.90Al0.10)Σ6.00O18] (BO3)3 V(OH)3 W[(OH)0.55F0.05O0.40]Σ1.00
End-member formula and relation to other species
The composition of the sample is consistent with a hydroxy-tourmaline belonging to the calcic group (Henry et al., Reference Henry, Novák, Hawthorne, Ertl, Dutrow, Uher and Pezzotta2011): it is Ca-dominant at the X position of the tourmaline general formula and hydroxy-dominant at W with (OH)– > O2–. As divalent-cations (with Mg) are the dominant Y-constituent, formula electroneutrality requires a Z-total charge = +17 in the end-member formula: CaMg3(Z6)Σ17+(Si6O18)(BO3)3(OH)3(OH). The unique charge arrangement compatible with the Z-constituents is Z(3+52+), which correspond to the atomic arrangement Z(Al3+5Mg2+). Therefore, the uvite end-member formula is CaMg3(Al5Mg)(Si6O18)(BO3)3(OH)3(OH). As no tourmalines are currently approved with this composition, it can be classified as a new species.
Uvite is related to fluor-uvite, ideally CaMg3(Al5Mg)(Si6O18)(BO3)3(OH)3F, and feruvite, ideally CaFe2+ 3(Al5Mg)(Si6O18)(BO3)3(OH)3(OH), by the homovalent substitutions W(OH)– ↔ WF– and YMg2+ ↔ YFe2+, respectively. Uvite is also related to magnesio-lucchesiite by the heterovalent substitution ZMg2+ + W(OH)– ↔ ZAl3+ + WO2–. The properties of these four tourmalines are compared in Table 9.
Table 9. Comparative data for uvite, fluor-uvite, magnesio-lucchesiite and feruvite.*

* Notes: The pleochroism reported in Grice and Robinson (Reference Grice and Robinson1989) is anomalous. All other tourmalines reported so far in literature display a reverse pleochroic scheme with O > E.
Calcic tourmalines from the thermal aureole of the Monte Capanne intrusion at San Piero in Campo: historical background and genetic inferences
To date, eleven tourmaline species have been identified from Elba Island. The origin of these tourmalines is both pegmatitic (elbaite, fluor-elbaite, schorl, foitite, rossmanite, tsilaisite, fluor-tsilaisite and celleriite) and non-pegmatitic related to basic hornfels which are mostly meta-serpentinites more or less altered by late-stage fluids (Ca-rich dravite, magnesio-lucchesiite and uvite) (e.g. Dini and Pezzotta, Reference Dini and Pezzotta2021). Among them, elbaite (Vernadsky, Reference Vernadsky1914), tsilaisite (Bosi et al., Reference Bosi, Skogby, Agrosì and Scandale2012), fluor-tsilaisite (Bosi et al., Reference Bosi, Andreozzi, Agrosì and Scandale2015b), magnesio-lucchesiite (Scribner et al., Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021), celleriite (Bosi et al., Reference Bosi, Pezzotta, Altieri, Andreozzi, Ballirano, Tempesta, Cempírek, Škoda, Filip, Čopjaková, Novák, Kampf, Scribner, Groat and Evans2022) and uvite (this study) have Elba Island as type locality. Uvite is the second calcic tourmaline, after magnesio-lucchesiite, discovered in the San Piero in Campo area. This locality has been well known since the late 18th Century for the occurrence of multi-coloured tourmaline specimens related to the presence of gem-bearing pegmatites (Pezzotta, Reference Pezzotta2021). The presence of black tourmaline in metabasite from Elba Island has been known since the 1820s (e.g. Soret, Reference Soret1822) and further descriptions were given by vom Rath (Reference Rath1870), D'Achiardi (Reference D'Achiardi1873), Viola and Ferrari (Reference Viola and Ferrari1911) and Millosevich (Reference Millosevich1914).
In metabasite from San Piero in Campo, calcic tourmalines (uvite and magnesio-lucchesiite) occur in the southeastern sector of the area, where metaserpentinite is in contact with intrusive rocks and are characterised by small vein systems filled by black-to-brown tourmalines. Veins are up to 2–3 cm thick and up to 1 m long. Their origin may be related to the influx of B-rich fluids released by the nearby pegmatite veins or leucogranitic and aplitic bodies during their crystallisation. The interaction between these B-rich fluids and (Ca/Mg)-rich metaserpentinites is consistent with the genesis of (Ca/Mg)-rich tourmalines. The compositions and the nature of the country rocks seem to control the crystal-chemistry of the calcic tourmalines from San Piero in Campo. Indeed, uvite has been identified in veins from the Facciatoia locality, hosted in deeply hydrothermalised metaserpentinite crosscut by magnesite + dolomite veins, nearby an LCT-pegmatite and a lens of Santa'Andrea facies monzogranite (Farina et al., Reference Farina, Dini, Innocenti, Rocchi and Westerman2010).
In the same area, magnesio-lucchesiite was identified by Scribner et al. (Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) in veins along fractures of a spinel-bearing rock occurring some hundreds of metres south-west of Facciatoia (San Rocco locality). Such veins contain tourmaline extremely enriched in Al (~7.7 apfu) with the spinel phase corresponding to Fe-bearing spinel, (Mg0.7Fe2+0.3)Σ1.0(Al1.9Fe3+0.1)Σ2.0O4, as determined by energy dispersive spectroscopy analyses. Scribner et al. (Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) identified in San Rocco a second occurrence of a solid solution between magnesio-lucchesiite and uvite relatively depleted in Al (~5.6 apfu), in veins along fractures within a basic hornfels derived by contact metamorphism of serpentinite.
The plot of Mgtot versus WO2– of the single spot analyses of the present uvite and the above-mentioned magnesio-lucchesiite samples show the occurrence of a solid-solution between these minerals (Fig. 5). The negative relation between Mgtot and WO2– is consistent in the substitution mechanism ZMg2+ + W(OH)– ↔ ZAl3+ + WO2–. Note that Mgtot was preferred to ZMg in Fig. 5 to remove issues of uncertainty associated with Mg and Al order–disorder over the Y and Z sites. In conclusion, the solid-solution between uvite and magnesio-lucchesiite is documented and shows the extreme sensitivity of the tourmaline-supergroup minerals to record subtle geochemical changes in the environment of crystallisation.

Fig. 5. The plot of Mgtot versus WO2– showing the occurrence of a solid-solution between uvite and magnesio-lucchesiite according to the substitution ZMg2+ + W(OH)– ↔ ZAl3+ + WO2– (see text). Data are single spot analyses of the uvite sample from this study (spots = 15) and magnesio-lucchesiite samples from Scribner et al. (Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) (Samples S1 and S2 from Elba Island, spots = 16 and 7, respectively; sample CAN from Canada, spots = 2).
Environments of uvite formation
High Ca and low F and Al contents are essential for stabilisation of uvite in contrast to other tourmaline species such as dravite, fluor-uvite and magnesio-lucchesiite. These specific conditions therefore require both specific host lithology and fluid composition.
Uvite occurrence in mafic rocks, similar to the type locality, was noted by Scribner et al. (Reference Scribner, Groat and Cempírek2018) who described uvite associated with Fe-rich dravite and Mg- and Ti-rich feruvite (and magnesio-lucchesiite; Scribner et al., Reference Scribner, Cempírek, Groat, Evans, Biagioni, Bosi, Dini, Hålenius, Orlandi and Pasero2021) in metasomatically altered lamprophyre dykes. The tourmaline formed by replacement of magmatic Ca-amphibole (actinolite to magnesio-hornblende) and its specific composition was constrained by the high Ca and low Al contents in the system during the metasomatic reaction.
Although tourmaline from calc-dolomite marbles is commonly described as ‘uvite’, these tourmalines typically contain high F and/or Na contents resulting in formation of fluor-uvite–magnesio-lucchesiite or dravite–oxy-dravite–magnesio-foitite solid solutions (e.g. Bačík et al., Reference Bačík, Uher, Cempírek and Vaculovič2012; Krmíček et al., Reference Krmíček, Novák, Trumbull, Cempírek and Houzar2021; Dutrow and Henry, Reference Dutrow and Henry2021). The occurrence of uvite with fluor-uvite in pockets of Portage-du-Fort marble, Québec, Canada, is a notable exception (Belley et al., Reference Belley, Grice, Fayek, Kowalski and Grew2014); uvite with significant WO contents (ca. 0.3–0.45 apfu; recalculation of original data to electroneutral formulae) forms mainly as a late retrograde mineral replacing serendibite in generations 5 and 6 in Belley et al. (Reference Belley, Grice, Fayek, Kowalski and Grew2014). Its association with fluor-uvite and fluor-dravite (generation 4) indicates that it formed from a F- and Na-depleted fluid at the final stages of retrograde tourmaline crystallisation.
In addition to specific Ca- and Mg-rich, Al-depleted environments such as hydrothermal veins in metaserpentinites (type locality) or metasomatically altered lamprophyres (Scribner et al., Reference Scribner, Groat and Cempírek2018), uvite may also form the fluor-uvite assemblages as a result of gradual change of fluid composition or local fluctuation in F content. The different environments and conditions that favour crystallisation of uvite show that calcic tourmalines may serve as an effective petrogenetic probe for monitoring fluid composition in hydrothermal and metamorphic systems.
Acknowledgments
Chemical analyses were done with the kind assistance of M. Serracino to whom the authors express their gratitude. F.B. acknowledges funding by Sapienza University of Rome (Prog. Università 2020). C.B. and F.B. acknowledges funding by the Ministero dell'Università e della Ricerca through the project PRIN 2020 “HYDROX – HYDRous- vs OXo-components in minerals: adding new pieces to the Earth's H2O cycle puzzle”, prot. 2020WYL4NY. F.C.H. acknowledges a Discovery Grant from the Natural Sciences and Engineering Council of Canada. Comments by the Structural Editor (P. Leverett) and reviewers (Vincent van Hinsberg and Oleg Vereshchagin) are very much appreciated.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2022.54
Competing interests
The authors declare none.

















 Open access
Open access