


Overview of copper homeostasis in the central nervous system (CNS)
Copper is a trace element that is essential for the functioning of numerous metalloenzymes due to its ability to readily shift between two oxidation states: reduced [Cu1+, Cu(I)] and oxidised [Cu2+, Cu(II)] (previously called cuprous and cupric, respectively); allowing either donation or acceptance of electrons in redox reactions. While copper is important for proper cellular physiology in all tissues (Ref. Reference Grubman and White1), it plays a crucial role in cellular functions that are required by the CNS, such as mitochondrial respiration, neurotransmission, iron metabolism, angiogenesis and free radical detoxification. In line with this, the average concentration of copper ions in the brain (80 µM) is notably higher than in peripheral tissues (kidney and lung, 30 µM; muscle, 10 µM) or blood (16 µM) (Ref. Reference Hamilton, Minski and Cleary2). For these reasons, absorption, transport and delivery of copper into the CNS, as well as copper homeostasis within brain cells are critical and tightly regulated by a number of copper-binding proteins and copper chaperones, as illustrated in Figure 1.

Figure 1. Schematic diagram of copper homeostasis, delivery and function in the CNS. Dietary copper must be reduced before being absorbed by intestinal enterocytes. As in most cell types, copper enters the enterocyte via the CTR1 transporter and is secreted through the trans-Golgi network and ATP7A. When copper reaches the blood, albumin and, to a much lesser extent, α2-macroglobulin transport it to the liver. Copper is absorbed into the hepatocyte and incorporated to CP. Copper-bound CP is secreted back to circulation through the hepatocyte TGN by ATP7B. The binding of copper to CP has a dual function: copper transportation throughout the body and iron oxidation, which is essential for the incorporation of ferric ion to transferrin and overall iron homeostasis. Copper passes through the BBB by active transport mediated by the combined action of CTR1 and ATP7A in brain capillaries. Once in the CNS parenchyma, copper is taken up into brain cells by CTR1 and likely by PrP, where it is incorporated as an active cofactor to key enzymes that are important for specific brain functions. In short, copper may bind SOD1 via transfer from CCS, and may also bind PARK7, which are crucial for maintaining proper protective mechanisms against free radical and metal toxicities. In addition, copper may be transported by COX17 to the intermembrane space of mitochondria, where in association with either COX11 or SCO1 and SCO2, it leads to the metalation of COX1 and COX2 and subsequent activation of complex IV. Beyond acting as an enzymatic cofactor, copper must be cleared from brain cells. This occurs, as in other cell types, through the transfer of copper to ATOX1, which delivers copper to ATP7A/B within the TGN, promoting the excretion of copper. Excretion requires the transport of copper into vesicles, a process mediated by the copper binding protein COMMD1. Lastly, copper might be stored intracellularly either within lysosome-like compartments via CTR2 or within the cytosol bound to copper binding MTs. MTs not only participate in the storage of intracellular excess of copper in a safe compartment but also provide chelation-dependent detoxification of free radicals and heavy/xenobiotic metals. ATP7A, copper-transporting ATPase 1; ATP7B, copper-transporting ATPase 2; ATOX1, copper transport protein ATOX1; BBB, blood-brain barrier; CCS, copper chaperone for superoxide dismutase; CNS, central nervous system; CP, ceruloplasmin; CTR1, high affinity copper uptake protein 1; CTR2, probable low affinity copper uptake protein 2; COMMD1, COMM domain-containing protein 1; COX1, cytochrome c oxidase subunit 1; COX2, cytochrome c oxidase subunit 2; COX11, cytochrome c oxidase assembly protein 11; COX17, cytochrome c oxidase copper chaperone; MTs, metallothioneins; PARK7, protein deglycase DJ-1; PrP, major prion protein; SCO1, protein SCO1 homolog, mitochondrial; SCO2, protein SCO2 homolog, mitochondrial; SOD1, superoxide dismutase [Cu-Zn].
The absorption of dietary copper occurs within the lumen of the small intestine after its reduction to Cu(I) by the duodenal cupric reductases cytochrome b (DCYTB) and/or STEAP2. Cu(I) is then taken up through the apical membrane of intestinal enterocytes, primarily by the copper transporter 1 (CTR1). In the cytosol, Cu(I) may be either stored in intracellular vesicles or transported to the trans-Golgi network (TGN) by the copper transporting ATPase (Cu-ATPase), ATP7A, from where it is finally exported from enterocytes to the blood (Ref. Reference Kim, Nevitt and Thiele3). Once in the blood, copper is primarily bound to albumin and to a much lesser extent to α2-macroglobulin for transport to the liver. Copper is absorbed into hepatocytes (through CTR1) where it is incorporated into a soluble ferroxidase called ceruloplasmin (CP). Copper-bound CP reaches circulation after being exported from the hepatocyte TGN by the action of another Cu-ATPase, ATP7B (Ref. Reference Roberts and Sarkar4). The binding of copper to CP has a dual function: copper transportation throughout the body and iron oxidation, which is essential for the binding of ferric ion to transferrin and overall iron homeostasis (Ref. Reference Hellman and Gitlin5).
Copper enters the CNS through the blood-brain barrier by active transport, mediated predominantly by the combined action of CTR1 and ATP7A in brain capillaries. Once in the brain parenchyma, copper is incorporated by brain cells, again mediated by CTR1, where it functions as an active cofactor to key enzymes that are important for specific brain functions. Indeed, mutations affecting copper homeostasis or copper-dependent proteins result in a range of diseases that are mainly characterised by neurodegenerative processes, as described in more detail below. In the CNS, it has been proposed that the prion protein (PrP), a plasma membrane-anchored protein that binds copper ions with high specificity, serves as a receptor for cellular uptake and/or efflux of copper (Ref. Reference Westergard, Christensen and Harris6). The role of PrP in copper trafficking has been supported by the correlation between PrP expression levels and copper content of cells (Ref. Reference Herms7); on the other hand, copper binding to PrP may also play a crucial role in the regulation of its physiological activity.
Once inside brain cells, copper may bind to the copper chaperone for superoxide dismutase, CCS, which activates cytosolic copper/zinc-dependent superoxide dismutase (SOD1) by inserting a copper ion and introducing a disulphide bond in the enzyme (Ref. Reference Culotta8). Copper may also bind to the protein deglycase DJ-1, also known as PARK7 (Ref. Reference Bjorkblom9). The activation of SOD1 and PARK7 by copper is crucial for maintaining proper protective mechanisms against free radical and metal toxicities. In addition, copper may be transported by cytochrome c oxidase copper chaperone protein (COX17) to mitochondria intermembrane space, where it serves the role of presenting bioavailable copper to the cytochrome c oxidase subunits 1 and 2 (SCO1/2) and cytochrome c oxidase assembly protein (COX11). COX11 and SCO1/2 lead to the metalation and activation of the complex IV (cytochrome c oxidase) through transferring copper to its catalytic core (subunits 1 [COX1] and 2 [COX2] respectively), responsible for electron transfer (Refs Reference Cobine, Pierrel and Winge10, Reference Horng11). The clearance of copper from brain cells occurs, as in other cell types, through the transfer of copper to the antioxidant 1 copper chaperone (ATOX1) protein, which delivers copper to ATP7A/B within the TGN, promoting the excretion of copper back to the brain parenchyma. This process requires the transport of copper into vesicles in the cell periphery by the COMMD1 protein (Ref. Reference Prohaska12). The oxidation of ATOX1 presumably results in a diminished delivery of copper to the secretory pathway, which likely reflects a functional link between redox balance and copper homeostasis (Ref. Reference Hatori13). Copper might not be excreted but instead stored either as intracellular copper pools in lysosome-like compartments via CTR2 (Ref. Reference van den Berghe14) or bound to copper-binding metallothioneins (MTs) in the cytosol (Ref. Reference Palacios, Atrian and Capdevila15). MTs, a family of low-molecular cysteine-rich proteins, are the main intracellular proteins that bind copper and zinc under physiological conditions (Ref. Reference Palmiter16). They not only participate in the intracellular storage of excess copper in a safe compartment but also provide chelation-dependent detoxification of free radicals and heavy/xenobiotic metals (Ref. Reference Ruttkay-Nedecky17).
Copper dysregulation in neurological disease
Copper deficiency compromises the proper functioning of cellular processes that are extremely important for tissues with high metabolic rates, such as the CNS and liver. Copper in the free-state is highly toxic to the CNS, since it can readily interact with oxygen to produce extremely damaging free hydroxyl radicals that overwhelm endogenous antioxidant systems. Therefore, a tight regulation of copper localisation and availability within the normal physiological range is of key importance in the CNS. As proof of this, genetic defects associated with altered copper homeostasis are the cause of severe neurological diseases. For instance, pathological mutations in the ATP7A and ATP7B genes, which encode two Cu-ATPases required for maintaining adequate copper levels within body tissues, are known to be involved in various brain pathologies.
Neurological disorders associated with defects in copper transporting ATPases
On the one hand, mutations in ATP7A are associated with Menkes disease (MNKD) (Refs Reference Tumer18, Reference Moller19) and distal spinal muscular atrophy, X-linked, 3 (DSMAX3) (Ref. Reference Kennerson20), both of which are characterised by generalised copper deficiency. MNKD results in a progressive and focal cerebral and cerebellar neurodegeneration with connective-tissue disturbances reflected in early growth retardation, peculiar hair, hypopigmentation, cutis laxa, vascular complications and death in early childhood; a mild form of the disease has also been described, causing cerebellar ataxia and moderate developmental delay. Two mutations have been described to cause DSMAX3, a neuromuscular disorder, which is characterised by motor neuropathy and selective degeneration of motor neurons (MNs) in the anterior horn of the spinal cord (SC) without sensory deficit in the posterior horn, and starts with weakness and wasting of distal muscles of the lower limbs and extends to the distal upper limbs.
On the other hand, defective function of ATP7B, leading to copper accumulation in the liver, kidney and CNS, causes Wilson disease (WD). More than 500 homozygous or heterozygous mutations have been associated with WD (Ref. Reference Lv21), a disorder that manifests with cirrhosis and a wide variety of neurological and psychiatric symptoms, including parkinsonism, ataxia, frontal lobe dementia, dystonia, seizures, migraines, depression, anxiety and psychosis. Further, aceruloplasminemia is caused by mutations in the CP gene (Ref. Reference Harris22), and although it is not associated exclusively with copper accumulation but also with iron deposition in the CNS and visceral organs, its clinical features consist of retinal degeneration, diabetes mellitus and neurological disturbances reminiscent of those of WD.
Copper and prion diseases
PrP is associated with several neurodegenerative diseases known as transmissible spongiform encephalopathies or prion diseases, examples in humans including Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Straussler disease, Huntington disease-like type 1, kuru and spongiform encephalopathy with neuropsychiatric features, which can be subdivided into sporadic, infectious or dominantly inherited forms. PrP exhibits strong binding affinity to copper (Ref. Reference Thompsett23) but the functional implication of copper binding to PrP is not yet clear. It has long been believed that copper ions play a role in PrP structural conversion (Ref. Reference Yu24), and indeed recent findings have provided direct molecular proof of the mechanisms by which an excess of copper binding to the octapeptide-repeat region of PrP leads to the conformational transition from the native PrP towards the misfolded and neurotoxic form (Ref. Reference Yen25). Nevertheless, due to the likely role of PrP in copper uptake and efflux (Ref. Reference Westergard, Christensen and Harris6), we should not rule out the idea that impaired copper delivery and/or excretion to/from brain cells to brain cells by pathological PrP might be contributing to the pathogenic role of this protein in prion diseases.
Copper and Alzheimer's disease (AD)
Neurological disorders are not only caused by genetic variations in copper-handling proteins, but also by mutations in enzymes that require copper as a cofactor, highlighting the importance of adequate copper availability for CNS function. One of the most exemplary cases is the β-amyloid precursor protein (APP), which is genetically and pathologically associated with AD and cerebral amyloid angiopathy. APP is a neuronal receptor involved in several functions related to neurite growth, neuronal adhesion, axonogenesis and axonal transport, but which also exerts a notable function in oxidative stress through copper ion reduction (Ref. Reference Ruiz26). On the other hand, an excess of copper accelerates pathogenic APP processing, β-amyloid aggregation and neurotoxicity (Refs Reference White27, Reference Hane28).
Copper and Parkinson's disease (PD)
Another common copper-dependent protein is α-synuclein, the coding product of the SNCA gene, which is associated with both the pathology and aetiology of some hereditary forms of PD. Indeed, the SNCA mutation H50Q enhances α-synuclein aggregation and toxicity chiefly due to the impaired binding affinity of this mutant protein to copper ions (Ref. Reference Khalaf29). In addition, the deglycase enzyme PARK7, which is involved in the protection of mitochondria from metal toxicity and oxidative stress when bound to copper (Ref. Reference Bjorkblom9), is also linked to familiar early-onset PD (Ref. Reference Bonifati30) and it is present in pathological tau protein inclusions (Ref. Reference Rizzu31). Interestingly, the E163 K mutation in PARK7 affects several copper-mediated functions of this enzyme (Ref. Reference Ramsey and Giasson32), resulting in impaired cytoprotection activity against oxidative stress in particular. Beyond early-onset PD, this mutation was found to cause dementia and amyotrophic lateral sclerosis (ALS) in an Italian family (Ref. Reference Annesi33), reminiscent of the phenotype of the cases from the ALS/parkinsonism-dementia cluster on Guam. Recently, a novel mutation in exon 3 of PARK7 (Q45X) has also been associated with both Parkinsonism and ALS in a Turkish family (Ref. Reference Hanagasi34), although it is unclear whether this mutation alters PARK7 function in a copper-dependent or -independent manner.
Copper and myopathies
Although very uncommon, mutations in SCO1 and SCO2 cause deficiency in the copper-dependent COX1 mitochondrial protein as well as subsequent mitochondrial respiration defects, which generally result in multisystem disease and particularly in neuromyopathies. Mutant SCO2 is associated with fatal infantile cardioencephalomyopathy (Ref. Reference Papadopoulou35), a disorder characterised by hypotonia, developmental delay, hypertrophic cardiomyopathy and lactic acidosis. This disorder is also characterised by gliosis and neuronal loss in the basal ganglia, brainstem and SC, which is reminiscent of spinal muscular atrophy (Ref. Reference Tarnopolsky36). Mutations in SCO1 cause mitochondrial complex IV deficiency (Ref. Reference Valnot37), a disorder with heterogeneous clinical manifestations ranging from myopathy to hypertrophic cardiomyopathy, hepatomegaly, hypotonia, muscle weakness, developmental delay, delayed motor development and mental retardation, but also encephalomyopathies such as Leigh syndrome.
Copper and motor neuron disease
Most of the diseases associated with mutations in copper-dependent enzymes and copper handling proteins, primarily ATP7A, ATP7B and to some extent PARK7, SCO1 and SCO2, have common symptomatic features, namely, neuromuscular defects and motor symptoms (Table 1). Beyond that, the relevance of copper in MN maintenance and ultimately neuromuscular degeneration is evidenced by the strong genetic and pathological association of SOD1 with familial ALS. One of the most copper-dependent enzymes is the intracellular antioxidant SOD1, in that not only the CCS protein has adopted the specific role of delivering copper to SOD1 but also other proteins such as DJ-1, MTs or glutathione may overlap this function (Refs Reference Seagrave38–Reference Carroll41) in order to assure the proper antioxidant function of this enzyme. While the triggering of MN degeneration by pathogenic mutations in SOD1 highlight the involvement of copper in the aetiopathogenesis of ALS through impaired SOD1, other copper-dependent mechanisms are likely to be involved. We will now review all the disease mechanisms and therapeutic tools for ALS in which copper is closely involved.
Table 1. Copper-associated changes in disorders with MND-like signs

MND, motoneuron degeneration; ATP7A, copper-transporting ATPase 1; ATP7B, copper-transporting ATPase 2; PARK7, protein deglycase DJ-1; SCO1, protein SCO1 homolog, mitochondrial; SCO2, protein SCO2 homolog, mitochondrial; SOD1, superoxide dismutase [Cu-Zn]; MN, motoneurons; ALS, amyotrophic lateral sclerosis.
Mechanistic insights into the role of copper dysregulation in the pathophysiology of ALS
ALS is a severe neuromuscular disease characterised by a progressive loss of MNs that results in paralysis and death within 2–5 years after disease onset. To date, the only approved treatment for ALS is riluzole, an antiglutamatergic drug that provides a slight increase in patient survival but has no effect on muscle wasting. The aetiology of ALS is complex and enormously heterogeneous, with multiple mechanisms contributing to the selective loss of MNs, including oxidative stress, mitochondrial dysfunction, glutamate excitotoxicity, neuroinflammation, endoplasmic reticulum stress and protein aggregation (Ref. Reference Vucic, Rothstein and Kiernan42). We will now outline the involvement of impaired copper homeostasis in these ALS pathogenic mechanisms.
Copper homeostasis and SOD1 toxicity
Mutations in the SOD1 gene have been extensively studied, mostly using transgenic mice expressing human mutant SOD1 protein, the most widely used animal model of ALS to date. SOD1 is a ubiquitous enzyme localised in the cytoplasmic and mitochondrial intermembrane spaces, which functions as scavenger of superoxide radicals by converting them into molecular oxygen and hydrogen peroxide. The fundamental basis of SOD1 toxicity in ALS remains to be elucidated. Nonetheless, the fact that both ablation and overexpression of endogenous wild-type (WT) SOD1 result in mild or no MN degeneration (Refs Reference Reaume43–Reference Dal Canto and Gurney45) supports the hypothesis that mutant SOD1 species found in ALS patients acquire altered conformations and a subsequent toxic gain of function triggers perturbations of cellular homeostasis and ultimately causes selective death of MNs.
The role of copper in the toxic effect of mutant SOD1 is currently under study. SOD1 monomers can be found in three different forms, depending on their metal content: (1) fully metalated (holo-) SOD1, which is bound to one zinc and to one copper metal ion; (2) fully demetalated (apo-) SOD1; and (3) metal-deficient SOD1, which is only bound to one metal ion, either zinc or copper (Fig. 2). SOD1 in the fully metalated form is one of the most stable and soluble enzymes known, with a melting temperature above 90°C and a long half-life (Ref. Reference Rodriguez46). On the other hand, copper deficiency has been shown to accelerate aberrant hydrophobicity of both WT and mutant SOD1 due to partial protein unfolding, though interestingly this aberration can be reverted by the addition of Cu(II) (Refs Reference Lynch and Colon47, Reference Tiwari48). In vitro studies have repeatedly indicated that recombinant SOD1 mutants are copper deficient (Refs Reference Corson49–Reference Ratovitski51).
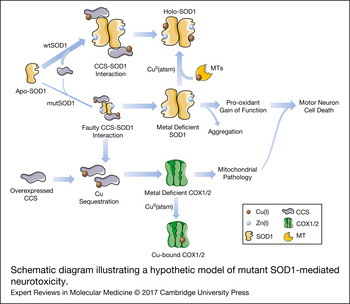
Figure 2. Schematic diagram illustrating a hypothetic model of mutant SOD1-mediated neurotoxicity. SOD1 can be found in a completely demetalated state (apo-SOD1), in a metal-deficient state, where SOD1 is only bound to one metal ion, either Zn(I) or Cu(I) or in a fully metalated state (holo-SOD1), where SOD1 is bound to both metals. The insertion of copper, which appears to occur mainly after SOD1 is bound to zinc and dimerized, is mediated by interaction with CCS. The interaction of CCS with mutant SOD1 is faulty, resulting in an accumulation of a less stabilized copper-deficient SOD1, which is prone to form aggregates and acquire a pro-oxidant and, consequently, toxic gain-of-function in motor neurons. The copper-deficient mutant SOD1 can be converted into the holo-form by the copper donor CuII(atsm), resulting in a decreased toxicity and delayed disease progression in animal models. Genetic or pharmacological upregulation of MTs, which may act as second copper source for SOD1, can also induce the conversion of metal-deficient SOD1 into the holo-form. In addition, the faulty interaction of CCS with mutant SOD1 leads to a situation in which copper remains sequestered in CCS. This situation may be aggravated by overexpressing CCS. Copper sequestration may result in a decreased delivery of copper to other enzymes, leaving for instance, COX1 and COX2 in a copper-deficient and dysfunctional state. This, in turn, would promote mitochondrial abnormalities that may strengthen mutant SOD1 toxicity. CCS, copper chaperone for superoxide dismutase; COX1, cytochrome c oxidase subunit 1; COX2, cytochrome c oxidase subunit 2; CuII(atsm), diacetyl-bis(4-methylthiosemicarbazonato)copperII; MTs, metallothioneins; mutSOD1, mutant superoxide dismutase [Cu-Zn]; wtSOD1, wild-type superoxide dismutase [Cu-Zn].
A recent study by Roberts et al. has provided deeper insights into the particular function of each metal content form of SOD1 in the context of ALS (Ref. Reference Roberts52). Several ALS-associated SOD1 mutations have been reported to alter protein structure and metal binding affinity. Notably, the G37R mutation does not seem to alter the overall SOD1 protein structure and it does not directly affect the metal-binding regions unlike, for example, H80R and D125H mutations (Ref. Reference Banci53). Nevertheless, Roberts et al. have found that more than 50% of the SOD1-G37R protein expressed in the SC of mutant mice is copper deficient, and that treatment with Cu(II) significantly decreases the levels of metal-deficient SOD1 forms while increasing the fully metalated SOD1 pool. Further studies have shown that mutant SOD1 in aggregates within cultured cells or within SCs of transgenic SOD1 mice, regardless of the mutants’ ability to bind copper, has a low copper content (Refs Reference Zetterstrom54–Reference Bourassa56) and that the degree of copper deficiency in mutant SOD1 aggregates is proportional to the clinical severity of the ALS (Ref. Reference Pratt57).
In combination with these studies, the aforementioned work of Roberts et al. (Ref. Reference Roberts52) refines the previous ALS mechanistic model of SOD1 mutant mice, by proposing that levels of metal-deficient SOD1 forms are better indicators of disease progression than the levels of total mutant SOD1 protein. On this basis, it is likely that MN death is caused by direct toxicity of the metal-deficient SOD1 proteins and therapeutic strategies should seek to reduce this pool through either metal chelation, to obtain apo-SOD1, which would be more readily degraded potentially mitigating the toxic effects of mutant SOD1, or better still, the addition of metal ions, to obtain holo-SOD1, which would improve stabilisation of the mutant protein into a non-toxic form. Remarkably, this model accomplishes the integration of seemly opposite findings from different groups into one comprehensive model. The importance of the work carried out by Roberts et al. lies in the insight it provides into SOD1-related toxic mechanisms in ALS (Ref. Reference Roberts52), with the demonstration that addition of copper either through CuII(atsm) or by overexpression of the copper transporter CTR1 has a significant therapeutic effect on SOD1-G37R mice.
Nevertheless, in order to test this model, the effects of zinc chelation should be determined in SOD1G37R or other copper-deficient ALS models. Further, the specific metal content of G37R and other mutant SOD1 proteins should be characterised more thoroughly in human in vitro systems, similar to the one previously described in CHO-K1 cells (Ref. Reference Bourassa56). In addition, in view of the importance of glial cells in ALS development, it would be interesting to analyse the specific distribution of metal-deficient mutant SOD1 in the different cell types, within the SC. Furthermore, it has to be considered that these findings were obtained mainly from animal models or cell lines, such as CHO, which do not necessarily accurately reflect human disease, and hence it is crucial to perform similar analysis in human samples and/or in newly developed induced-pluripotent stem cells (iPSC)-derived MN culture systems. Finally, the role of SOD1 aggregates in ALS pathology remains to be clarified, but since oral treatment with CuII(atsm) does not decrease levels of misfolded SOD1 (Ref. Reference Roberts52), it is very likely that aggregates make a marginal contribution to decreased neuronal function. Indeed, in vitro experimental data have shown that the toxicity of mutant SOD1 protein is dramatically enhanced after increasing its solubility, which implies that the reduced solubility of mutant SOD1 proteins achieved through aggregation may be a self-protective response (Ref. Reference Brotherton, Li and Glass58).
Beyond its prominent role in redox homeostasis, SOD1 also has an important function as a copper buffer within the cell (Ref. Reference Culotta59), and hence, impaired cellular functions that are fully or partially dependent on copper availability may also be contributing to the toxic effects of mutant SOD1 proteins. Therefore, the contribution of other copper-dependent enzymes in the pathogenic cascade of SOD1 toxicity, as a consequence of disrupted copper homeostasis, should be included in the model proposed by Robert and colleagues (Fig. 2). In relation to this, alterations in copper influx/efflux have been evidenced in the SOD1-G93A mice. In particular, elevated concentrations of copper in this mutant SOD1 model have been reported early at pre-symptomatic phases within the SC and skeletal muscles, and this condition aggravates as disease progresses (Refs Reference Tokuda, Okawa and Ono60, Reference Enge61), suggesting that copper dyshomeostasis might facilitate the development of ALS. Further, studies in mice with genetic modifications in copper-handling enzymes have confirmed the putative role of copper homeostasis in SOD1 toxicity. For example, the overexpression of CCS in the SOD1-G93A mice, although promoting the conversion of apo-SOD1 into the more stable holo form, led to an accelerated pathology and disease progression (Ref. Reference Son62) that was abolished by treatment with a complex that delivers copper (Ref. Reference Williams63). In another study, the ablation of CCS in three different lines overexpressing distinct SOD1 mutations induced a significant reduction in copper-loaded SOD1 without affecting incorporation of the metal into other copper-dependent enzymes. Interestingly, the disease onset and progression were not modified (Ref. Reference Subramaniam64), suggesting that copper transferred by CCS is not involved in SOD1 toxicity (Ref. Reference Beckman65). Conversely, the copper homeostasis in SCs from mutant SOD1 mice by genetic or drug-induced overexpression of different types of MTs, important copper chaperones acting against oxidative and metal toxicity, has shown significant positive effects on pathological features and motor symptoms without influencing SOD1 activity (Refs Reference Hashimoto66–Reference Tokuda68). These findings might be explained by the fact that MTs are crucial for intracellular copper storage (Ref. Reference Tapia69) and can even act as a secondary source of copper for SOD1 or other enzymes (Refs Reference Seagrave38, Reference Liu39). In relation to this, copper delivery to SOD1 and subsequent SOD1 stabilisation in CCS-deficient cells could be compensated for by the pool of intracellular MTs. An excess of copper-loaded CCS as a consequence of either overexpression of the CCS protein or faulty interaction and transfer of copper from CCS to mutant SOD1 might leave other enzymes, such as cytochrome c oxidase subunits COX1 and COX2, in a copper-deficient state, thereby inducing or potentiating the neurotoxic effects of mutated SOD1 (Fig. 2). Indeed, the researchers who developed double transgenic CCS/SOD1-G93A mice have already reported deficient activation of mitochondrial complex IV (in terms of decreased protein levels of subunits COX1 and COX5b, reduced subunit assembly and diminished cytochrome c oxidase activity) in this model as a part of the mutant SOD1-induced mitochondrial dysfunction (Refs Reference Son62, Reference Son70). Nevertheless, as authors do not report mRNA levels of these subunits, it is not known whether reduced complex IV activity is primarily because of compromised expression of COX1 and COX5b subunits in this double transgenic mouse. In contrast, copper-deficient COX1 might perturb the assembly of complex IV subunits (Ref. Reference Fontanesi71), and hence be the cause of the reported reduction in both protein levels and complex IV activity. Additionally, a missense mutation in CCS, which abrogates the interaction of CCS with SOD1 (Ref. Reference Wright, Antonyuk and Hasnain72), has been reported in an individual from a Turkish family with delayed psychomotor development, brain atrophy, hypomyelination, muscular hypotonia and, interestingly, low mitochondrial complex IV activity on muscle biopsy (Ref. Reference Huppke73).
Future research should focus on exploring novel strategies to prevent SOD1 toxicity mediated by metal deficiency. Such strategies might also be useful for other disorders with abnormal metal homeostasis, such as PD, Huntington's disease and stroke. Further insights into SOD1 toxic mechanisms can lead to novel therapeutic avenues for future research concerning ALS as well as other neurological diseases.
Copper homeostasis and mitochondrial dysfunction
Mitochondrial dysfunction is a major hallmark of MN degeneration in ALS (Ref. Reference Muyderman and Chen74). Specific alterations in mitochondrial morphology, such as swelling and vacuolisation, along with metabolic deficits in the activities of the respiratory chain complexes have been extensively detected in postmortem tissues and in lymphocytes from sporadic ALS patients (Refs Reference Carri, D'Ambrosi and Cozzolino75, Reference Wiedemann76), as well as in SOD1 transgenic mouse and cell models (Ref. Reference Son and Elliott77). Experimental studies have even indicated that such mitochondrial defects occur at presymptomatic stages before the onset of MN loss (Refs Reference Kong and Xu78–Reference Damiano80). These defects are generally associated and interrelated with both bioenergetic failure and increased oxidative damage.
In the literature, there are relatively few data directly related to the role of copper deficiency in mitochondrial dysfunction in ALS. In one of the aforementioned studies, the overexpression of CCS in a mutant SOD1 mouse augmented the incorporation of copper into SOD1 as well as the severity of the disease. Interestingly, the disease phenotype in this double transgenic mouse was not related to toxicity due to aggregated SOD1 but instead to severe mitochondrial pathology in the SC (Ref. Reference Son62). Beyond this study, some evidence of ALS-related changes in certain copper-dependent enzymes with prominent roles in mitochondrial function support the view that mitochondrial copper is involved in the pathophysiology of ALS. For example, mutant PARK7 is a common trigger of neuronal death through a mechanism affecting mitochondrial health and function (Ref. Reference Chaturvedi and Flint Beal81). Although the function of the protein product of this gene, DJ-1, has not been fully characterised, it has been established that it contributes to the maintenance of mitochondrial function by regulating mitophagy and cell tolerance to reactive oxygen species (Ref. Reference Wang82). Mutations in PARK7 often compromise the copper-binding ability of DJ-1, thereby underlining the importance of copper-bound DJ-1 in the correct physiology of mitochondria. Although PARK7 mutations are strongly associated with PD, and only in certain cases with the ALS-dementia-parkinsonism complex, the expression of DJ-1 is altered in sporadic ALS patients and mouse models (Refs Reference Lev83–Reference Knippenberg85). Indeed, the genetic ablation of DJ-1 in a mutant SOD1 mouse aggravated MN loss, gliosis and oxidative damage in the SC and shortened mutant mouse lifespan through an Nrf2-dependent mechanism (Ref. Reference Lev86). Nrf2 is a transcription factor that has been found to master regulate the oxidative response by modulating the expression of genes promoting mitochondrial biogenesis and maintenance (Ref. Reference Dinkova-Kostova and Abramov87), among others. The same study also observed neuroprotective effects of a DJ-1-derived peptide in an MN cell model of glutamate-induced toxicity. Overexpression of DJ-1 in mutant SOD1-expressing MN cultures has also been reported to interact physically with mutant SOD1 to reduce its toxicity (Ref. Reference Yamashita84), likely to the potential function of DJ-1 as an alternative chaperone to transfer copper ions to SOD1 (Ref. Reference Girotto40). Another protein with primary role in mitochondrial function whose activity relies strongly on copper levels is complex IV subunits COX1 and COX2. Complex IV activity has been found to be low not only in postmortem SCs of ALS patients (Refs Reference Wiedemann76, Reference Fujita88, Reference Borthwick89) and animal models (Refs Reference Igoudjil90–Reference Kirkinezos92), but also in muscle biopsies from individuals with sporadic ALS (Refs Reference Russell93, Reference Crugnola94) or SOD1 familial ALS (Ref. Reference Corti95); moreover, in blood samples from living patients, the activity was found to be correlated with clinical staging of disease severity (Ref. Reference Ehinger96). Though some other studies have not found any alteration in complex IV activity in transgenic mice (Ref. Reference Bendotti97) or blood from patients (Ref. Reference Gluck, Thomas and Sivak98), most available evidence supports the view that there are generalised ubiquitous reductions in complex IV activity in ALS. Interestingly, the accelerated ALS mouse created by Son et al., which overexpresses mutant SOD1-G93A together with CCS, displayed significantly low complex IV activity despite normal complex I, II, III and V function (Ref. Reference Son70), again manifesting the crucial involvement of copper dyshomeostasis, likely with the participation of pathological SOD1, in the biochemical abnormalities of mitochondria observed in ALS. In any case, apart from one old scientific publication from the 1960s that reported low cytochrome c oxidase activity in MNs of lambs with brain copper deficiency and ataxic swayback lesions (Ref. Reference Fell, Mills and BOYNE99), the potential link between copper and complex IV activity in MN degeneration remains to be elucidated.
Copper homeostasis and TDP-43 pathology
Neuronal and glial cytoplasmic deposition of aberrant misfolded TDP-43 in co-localisation with p62 and ubiquitin is a common pathological hallmark in SCs, hippocampi and cerebella of ALS patients, except in familial SOD1 and FUS forms, which barely account for 1–2% of all cases (Ref. Reference Blokhuis100). The mechanisms leading to TDP-43 mislocalisation and accumulation in the cytoplasm are not yet fully understood. It has been observed that ALS patients with mutations in the 3'UTR region of its encoding gene (TARDBP) show overexpressed levels of TDP-43. This deregulated production of TDP-43 might underlie its mislocalisation and accumulation in the cytoplasm (Ref. Reference Neumann101).
On the other hand, elevated levels of WT or mutant TDP-43 do not always give rise to insoluble cytoplasmic inclusions, as found in other non-CNS tissues, suggesting the involvement of defective autophagy-dependent clearance mechanisms or the presence of pro-aggregation agents, both contributing to the eventual deposition of pathological TDP-43. Indeed, the co-localisation of several ribonucleoproteins of RNA stress granules (SGs) with TDP-43 within the cytoplasmic inclusion suggests that these inclusions are remnants of SGs whose components could not be reused by the cell and remain as residual bodies (Ref. Reference Li102).
Although TDP-43 does not possess the ability to bind copper ions, it has been demonstrated that this metal may influence the aggregation state of TDP-43. For instance, the exogenous addition of reduced copper ions to the neuroblastoma SH-SY5Y cell line abolishes the paraquat-mediated formation of endogenous TDP-43 aggregates into SGs and subsequent cell death (Ref. Reference Parker103). The role of copper in protecting against TDP-43 accumulation has been described in terms of an ability of copper to inhibit prolonged ERK activation and subsequent abnormal ubiquitin-dependent proteasome degradation of TDP-43. Furthermore, the prevention of phosphorylated and fragmented TDP-43 aggregation by copper has also been confirmed in the mutant TDP-43 (A315 T) transgenic mouse (Ref. Reference Soon104).
Conversely, it has also been reported that the mutant TDP-43 (A315 T) can affect copper homeostasis. In particular, Dang et al. found that the overexpression of this mutant protein in the whole CNS disrupted copper homeostasis, increasing its content in the SC but not in the brain, resulting in motor symptoms but not cognitive deficits (Ref. Reference Dang105). This suggests a particular vulnerability of MNs to copper homeostasis mechanisms. In general, despite scarce experimental evidence of copper defects in relation to ALS-pathological TDP-43, the apparent cross-talk between these two pathogenic pathways is interesting and merits further investigation.
Targeting copper delivery to treat MN disease
Radiolabeled diacetyl-bis(4-methylthiosemicarbazonato)copperII, (CuII(atsm)) is an effective positron-emission tomography imaging agent for myocardial ischemia, hypoxic tumours and brain disorders with regionalised oxidative stress. CuII(atsm), which transports Cu(II) throughout the body and delivers it when it reaches oxidative tissues, has shown a therapeutic effect in several animal models of ALS and PD (Refs Reference Roberts52, Reference Williams63, Reference Soon104, Reference Hung106–Reference Hilton108). In particular, CuII(atsm) significantly improves locomotor function and increases mouse survival in SOD1-G93A, SOD1-G37R (Refs Reference Roberts52, Reference Soon104, Reference McAllum107, Reference Hilton108) and TDP-43 (A315 T) mouse models of ALS (Ref. Reference Soon104). In their work, Soon et al. showed that CuII(atsm) functions as an effective scavenger of peroxynitrite, and they proposed that this capability may underlie its beneficial therapeutic effect on ALS symptoms. In line with this, in animal models of PD, CuII(atsm) improved motor and cognition function and rescued cell loss within substantia nigra by reducing peroxynitrite-driven toxicity and promoting neuroprotection (Ref. Reference Hung106).
Roberts et al. focused on understanding the fundamental basis of mutant SOD1 toxicity in ALS and provided the first evidence that part of the therapeutic effects of CuII(atsm) in the SOD1-G37R mouse may be a consequence of a copper transfer from CuII(atsm) to the SOD1 protein. The authors demonstrated that orally administered CuII(atsm) is able to reach the CNS and transfer a Cu(II) ion to SOD1 in the SC, thus functioning as a copper donor. In a first set of experiments, they showed that CuII(atsm) treatment protects SOD1-G37R mice from the toxic effects of mutant SOD1, improving mouse locomotor function and survival. Furthermore, they found that CuII(atsm) has a neuroprotective effect on MNs in the SC, while decreasing inflammation and oxidative levels. Interestingly, the authors found that even though CuII(atsm) reduces disease progression, it significantly increases protein levels of mutant SOD1. The observation that oral CuII(atsm) treatment induces an increase of both mutant SOD1 protein and activity levels was further recapitulated in the SOD1-G93A mouse, with the particularity that these changes are specific for SC but absent in other peripheral tissues such as liver (Ref. Reference Hilton108), highlighting the CNS-selective effects of mutant SOD1 and response to this drug. The latter is in line with the limited capacity of CNS to respond to an increased requirement for endogenous copper when mutant SOD1 is overexpressed (Ref. Reference Hilton, White and Crouch109). Both evidences provide novel insights into the CNS-specific vulnerability to SOD1 toxicity in ALS and place copper as key element in this process.
The repeated findings that copper supplementation-based therapy significantly raises the levels of mutant SOD1 protein in SC disapprove previous claims that the severity of the mutant ALS mouse phenotype was proportional to levels of mutant SOD1 proteins. In addition, mutant SOD1 misfolding and aggregation have been commonly related to SOD1-mediated toxicity (Ref. Reference Vucic, Rothstein and Kiernan42). However, Roberts et al. did not observe any significant change in the levels of misfolded SOD1 proteins in SCs when SOD1-G37R mutant mice were treated with CuII(atsm). Conversely, they found that enhancing intracellular copper uptake by overexpressing the transporter CTR1 in the mutant SOD1 mouse reproduced the beneficial effects of CuII(atsm) in terms of survival and disease progression. In another study, the same group treated SOD1-G73R mice with ZnII(atsm) and reported similar improvements in locomotor function and survival of the mice (Ref. Reference McAllum110). The hypothesis that delivery of zinc to mutant SOD1 could be therapeutic follows the rationale that zinc metalation is also required for the formation of holo-SOD1 and plays even a more remarkable role than copper suppressing the aggregation of SOD1 during refolding (Ref. Reference Li111). However, this treatment did not increase overall zinc levels in the SC nor the zinc content of SOD1, while it did increase overall levels of copper and the copper content of SOD1. Authors demonstrated a transmetalation of ZnII(atsm) to the analogue CuII(atsm) in the presence of copper and concluded that the observed therapeutic effects for ZnII(atsm) may in fact be due to subsequent delivery of copper (Ref. Reference McAllum110).
In another study, CuII(atsm) was transdermally administered to the animal model with the most rapid progression ALS, the double transgenic mouse overexpressing both SOD1-G93A and CCS. CuII(atsm) extended survival from 30–50 days to 18 months and was able to rescue motor deficits when the treatment was given in symptomatic mice (Ref. Reference Williams63). Withdrawal of CuII(atsm) resulted in increased nitrotyrosine accumulation, pronounced astrogliosis and reduced mitochondrial complex IV cytochrome c oxidase activity in the SC. CuII(atsm) treatment induced a reduction in the amount of copper-deficient SOD1 in SOD1-G93A, which was exacerbated in the SOD1/CCS transgenic double mice. CuII(atsm) treatment in these mice also led to a marked accumulation of SOD1, but its withdrawal did not result in an accumulation of copper-deficient SOD1.
Overall, these findings lead to the conclusion that improving copper content represents a promising therapeutic strategy for treating ALS. However, it remains to be elucidated whether copper incorporation into mutant SOD1 is sufficient or whether the metalation of other copper-dependent enzymes, beyond metalation of SOD1, is also involved in the observed therapeutic effects of CuII(atsm). (Fig. 2).
In contrast to the idea of using copper-delivery therapies for ALS, there are a number of reports showing the beneficial effects of copper-chelating drugs on survival and disease progression in mutant SOD1 mouse models. In particular, studies by Tokuda et al. tested the copper chelator ammonium tetrathiomolybdate (TTM) as a strategy to remove the copper accumulation in SCs that is driven by mutant SOD1 proteins. They found that TTM treatment prolonged survival of both presymptomatic and symptomatic SOD1-G93A mice, and attenuated severity in terms of decreased MN loss and skeletal muscle atrophy. Interestingly, TTM is capable of removing copper ions from the copper-thiolate cluster of SOD1, resulting in a significant suppression of SOD1 activity and reduced aggregation of mutant forms (Refs Reference Tokuda112, Reference Tokuda113). Hence, copper-chelation approaches might likely promote the formation of demetalated mutant apo-SOD1, which would be more readily degraded (Ref. Reference Urushitani114) potentially mitigating the toxic effects of mutant SOD1. Although TTM is an approved metal chelating agent specific for copper that is used for the treatment of WD and pulmonary fibrosis, it has other important characteristics such as being hydrogen donor with strong antioxidant and cytoprotective properties (Ref. Reference Xu115). Other studies have reported beneficial outcomes in transgenic SOD1 mouse models when testing various different approaches to remove copper with trientine (Refs Reference Andreassen116–Reference Nagano118), d-penicillamine (Ref. Reference Hottinger119) or other lipophilic chelators (Refs Reference Petri120, Reference Azzouz121), though none of these chelating drugs are specific for copper (rather they chelate other heavy metal ions, such as lead, iron or mercury). Hence, experimental outcomes in preclinical trials with copper-chelating drugs could also be interpreted with the caution that their efficacy in ALS models might be likely dependent on the homeostasis of other metal ions or the regulation of other pathogenic mechanisms.
Concluding remarks and perspectives
The role of copper ions in the development and functions of the CNS is now unquestionable, because of the clinical evidence from numerous mutations in different genes involved in the homeostasis of this metal giving rise to heterogeneous clinical manifestations with a common denominator: neurodegeneration and neurological symptomatology. The involvement of copper homeostasis in the pathophysiology of ALS requires special attention because of the important role of copper as a cofactor in the regulation of SOD1 stabilisation and activity.
While several studies have recently identified the copper-deficient state of SOD1 as the principal mediator of mutant SOD1 toxicity, providing promising data on the therapeutic potential of copper to restore the copper-deficient state of SOD1, many questions remain to be addressed: (1) since copper-chelating therapies have also produced beneficial outcomes in animal models, is copper neurotoxicity actually related to the metal-deficient state of some important enzymes, such as SOD1, or to copper accumulation as a consequence of the latter, which might be interfering in important cellular pathways? (2) Since protein nitration represents a major cytotoxic pathway contributing to neurodegeneration in ALS and CuII(atsm) is both a copper donor and an anti-nitrosative agent, to what extent are the different effects responsible for the overall therapeutic benefit of this compound? (3) Since mutant SOD1 is the cause of a small percentage of ALS cases, though the aetiology, pathophysiology and proteinopathy of most ALS cases are SOD1-independent, to what extent is copper dyshomeostasis involved in the pathogenic cascade of TDP-43, FUS or perhaps in the formation of pathogenic C9ORF72 GGGGCCexp RNA foci? Further, how effective are copper-based therapies in the novel non-SOD1 genetic animal models currently being developed and characterised? (4) Which might be the most valuable tools for bringing significant novel insights into the mechanisms of copper dysregulation in ALS and/or for the screening of copper compounds? For instance, newly developed iPSC-derived MN cultures from sporadic/familial ALS patients or with CRISPR-Cas-mediated mutations in copper-handling genes? (5) What can we learn from studying the biology of copper MTs, since there is growing evidence that these proteins could be playing a key role in copper homeostasis, acting as a second copper source that regulates the correct functioning not only of SOD1 but also of other copper-dependent enzymes, which may be involved in the pathophysiology of ALS, such as COX1/2 or PARK7.
Therefore, the development of sensitive and high-resolution techniques for studying the chemistry, biochemistry and biology of copper, as well as the utilisation of animal/cellular models, which are relevant to the pathological process of the disease, are crucial to further investigate and consolidate our knowledge of the biological functions of copper in the pathophysiology of ALS and other neurodegenerative diseases.
Acknowledgements and funding
We thank Dr Virginia Arechavala for helpful comments. We also dedicate this work to all ALS patients and their relatives. This work was supported by grants from the Carlos III Health Institute (PI14/0219 to AVI; PI2016/04 to FGB), and the Basque Government (2016111091 to AVI, 2015111122 to FGB). GA holds a PhD fellowship from the Basque Government, HLF holds a PhD fellowship from the University of the Basque Country and AVI is a Ramón y Cajal Fellow.
Conflicts of interest
None.
Ethical standards
This review work did not involve human and/or animal experimentation.



