


INTRODUCTION
Leishmaniasis refers to a diverse group of diseases caused by Leishmania spp., protozoan parasites of the family of Trypanosomatidae. Leishmaniasis is geographically widespread because different Leishmania spp. are capable of adapting to different environmental circumstances. Leishmania spp. have established themselves in urban and rural settings, in deserts and forests and are transmitted by the bite of an infected sand fly. Humans exhibit visceral, cutaneous and mucocutaneous forms of infection [1]. The variability of the disease depends on the host's immune response and on the particular species of Leishmania [Reference Anstead2–Reference Meddeb-Garnaoui, Zrelli and Dellagi4].
Leishmaniasis is endemic in 98 countries worldwide, and more than 350 million people are exposed to infection [1]. It has been recently estimated that 0·5 million new cases of visceral leishmaniasis occur each year, with 50000 cases leading to death, and 1·5 million cutaneous leishmaniasis cases occur annually [1].
The diagnosis of leishmaniasis in humans is usually made by serological tests, in vitro cultivation and detection of the parasites in tissue aspirates [Reference Herwaldt5, Reference Singh and Sivakumar6]. In the last two decades, Polymerase chain reaction (PCR) assays have been used for more sensitive and accurate diagnosis of these diseases [Reference Cruz7, Reference Reithinger and Dujardin8]. However, all of these methodologies are time-consuming and require special, expensive equipment and a well-trained and experienced staff [Reference Mori and Notomi9]. These circumstances are lacking in developing countries, particularly in the areas where the disease is most widespread. Furthermore, in subclinical cases or when the number of parasites is low, such as in patients with visceral leishmaniasis, microscopy may be unsuccessful in detecting the disease. Taking these facts into consideration, there is a great demand for a technique that will enable a definite, simple, rapid and cost-effective diagnosis of leishmaniasis.
In recent years, a new molecular-based method, i.e. loop-mediated isothermal amplification (LAMP), has been successfully applied for the detection of other pathogens. LAMP promises to be a rapid, specific, sensitive and simple assay for pathogen detection and identification. LAMP can amplify DNA or RNA under isothermal conditions in 1 h [Reference Mori and Notomi9] and its popularity has rapidly grown for different applications and areas [Reference Karanis and Ongerth10]. LAMP has already been used for the diagnosis of several parasitic diseases, including malaria [Reference Pöschl11], cryptosporidiosis [Reference Inomata12, Reference Plutzer, Törökné and Karanis13], toxoplasmosis [Reference Sotiriadou and Karanis14], trypanosomiasis [Reference Thekisoe15] schistosomiasis [Reference Xu and Rong16].
The aim of the present study was to develop and evaluate a unified LAMP protocol in bench-scale experiments in vitro and to utilize it for the detection of a wide range of Leishmania spp. that cause cutaneous, mucocutaneous and visceral leishmaniasis. Two LAMP primer pairs for the amplification of Leishmania DNA were designed to provide improved coverage of globally diverse species together with high specificity as no other protozoan DNA was amplified.
MATERIALS AND METHODS
Origin of Leishmania DNA
The development of LAMP assays required DNA from Leishmania spp. Seven reference strains were cultivated under sterile conditions and incubated at 26°C. The Leishmania spp. that were used for cultivation were L. donovani (zymodeme MON-2, MHOM/IN/1996/THAK35), L. major LV39 (zymodeme MON-4, MRHO/SU/59/P), L. infantum L4 (zymodeme MON-1) (obtained from the Pasteur Institute, Athens, Greece) and L. major 230/119 (zymodeme MON-4), L. major 159/06, L. tropica 208/48 (zymodeme MON-60), L. tropica 101/65 (obtained from the Institute of Medical Parasitology, Koblenz, Germany).
Three types of media were prepared and used for the cultivation of the individual strains. L. major LV39 and L. donovani MHOM/IN/1996/THAK35 were cultivated in RPMI-1640 medium [Reference Tseveleki17], whereas L. infantum L4 was cultivated in DMEM. Leibovitz's medium was used for the cultures of L. major 230/119, L. major 159/06, L. tropica 208/48 and L. tropica 101/65 [Reference Dweik18]. Leishmania subcultures were generated by inoculating tubes containing fresh medium with parasites once a week under sterile conditions.
The DNA of the cultivated parasites was extracted by the phenol-chloroform extraction method [Reference Dweik18] and frozen at −20°C until use.
DNA from L. guyanensis (referred to by the WHO as MHOM/GF/79/LEM85), L. braziliensis (referred to by the WHO as MHOM/BR/00/LTB300), L. donovani DON-43, Trypanosoma evansi, T. brucei, Cryptosporidium parvum, Toxoplasma gondii, Giardia lamblia and Entamoeba histolytica were also used for conducting specificity tests.
Primer design
Two sets of four primers were designed to be specific for Leishmania spp. based on the 18S rRNA gene (Table 1, Figs 1 and 2). The sequence of the L. major rRNA-encoding gene on chromosome 27, from nt 989 660 to 9 918 63 was used. The primers were designed to detect L. donovani, L. major, L. infantum, L. braziliensis, L. guyanensis, L. tropica, L. mexicana, L. amazonensis, L. chagasi, L. sp. siamensis, L. naiffi, L. lainsoni and L. panamensis.

Fig. 1. LAMP primer set 1, targeting the Leishmania major rRNA-encoding gene on chromosome 27. The partial sequence of L. major and the location of four primers, FIP (F1c-F2), BIP (B1c-B2), F3, and B3 are shown. The arrows indicate the direction of the extension. The numbers on the left indicate the nucleotide position.

Fig. 2. LAMP primer set 2, targeting the Leishmania major rRNA-encoding gene on chromosome 27. The partial sequence of L. major and the location of four primers, FIP (F1c-F2), BIP (B1c-B2), F3, and B3 are shown. The arrows indicate the direction of the extension. The numbers on the left indicate the nucleotide position.
Table 1. Nucleotide sequences of LAMP primers (sets 1 and 2) designed for detection of Leishmania promastigotes

LAMP
The components of the reaction mixture were 1 μl primer mix (5 pmol F3, 5 pmol B3, 40 pmol FIP, 40 pmol BIP), 1 μl (8 U) Bst DNA polymerase, 2 μl DNA sample, 12·5 μl of 2x LAMP buffer containing 1·4 mm each of dNTP, 0·8 m betaine, 20 mm Tris–HCl (pH 8·8), 10 mm KCl, 10 mm (NH4)2SO4, 8 mm MgSO4 and 0·1% Tween 20. The mixture was tested with both primer sets, and the reactions were performed at temperatures ranging from 58°C to 70°C and for incubation times ranging from 50 to 120 min to determine the optimal amplification conditions. Visualization of LAMP products was performed by gel electrophoresis using 1·6% agarose gels stained with ethidium bromide (1 μg/μl) and run for 30 min at 100 V in an electrophoresis chamber. When the electrophoresis was complete, the gel was carefully transferred to a gel documentation system and visualized by UV transillumination.
Nested PCR amplification
For the PCR detection of Leishmania, the gene selected was the internal transcribed spacer-1 gene (ITS-1), which is part of the ribosomal RNA gene of Leishmania. The primers used were LITSRn (5′-CTG GAT CAT TTT CCG ATG-3′) and L5·8 s (5′-TGA TAC CAC TTA TCG CAC TT-3′) [Reference Dweik18]. The reaction mixture contained 5 μl of 10x PCR buffer (Qiagen, Germany), 2·5 mm each of dNTP (Fermentas, Canada), 10 pmol of each primer, 1 U HotStarTaq polymerase (Qiagen), 2·5% DMSO, distilled water and 5 μl DNA template. The conditions of the amplification in the thermal cycler were 95°C for 15 min, then 34 cycles of the following steps: denaturation at 95°C for 20 s, annealing at 53°C for 30 s and extension at 72°C for 60 s. After completion of 34 cycles, the reaction was subjected to final extension of 72°C for 6 min. The second PCR was performed under the same amplification conditions and with the same reaction mix as the first PCR, except that the DNA template was 1 μl PCR product from the first amplification. The PCR products were electrophoresed as described above.
Sensitivity
The sensitivity of the PCR assay was tested using samples containing the target DNA of L. major 230/119 with concentrations of DNA ranging from the DNA of 1 × 105 promastigotes/5 μl DNA template to the DNA of 1 promastigote/5 μl. Promastigotes were counted by haemacytometer prior to DNA extraction. A negative control of a sample containing distilled water instead of DNA was always included.
Specificity
To ensure that DNA from several Leishmania spp. and strains could be amplified, the specificity of the PCR assay was assessed using cultured reference strains of L. donovani MHOM/IN/1996/THAK35, L. infantum L4, L. major 159/06, and L. major LV39. Additionally, the DNA of various other parasites, including T. evansi, T. brucei, Toxoplasma gondii, C. parvum, G. lamblia and E. histolytica, was assayed to test the specificity of the PCR assay.
LAMP primer working conditions
The LAMP product was detected by agarose gel electrophoresis and exhibited a specific pattern of multiple bands. After examining all the cultivated reference strains, the optimal conditions for amplification were found to be 65°C and 85 min for primer set 1 and 63°C and 85 min for primer set 2. At the end of the reaction, the temperature was increased to 80°C for 5 min to terminate the reaction.
RESULTS
Testing LAMP primer set 1
The DNA from ten different Leishmania isolates as well as from other parasites was examined to test the specificity of primer set 1. All samples containing DNA from the Leishmania spp. (L. donovani THAK35, L. infantum L4, L. major LV39, L. donovani DON-43, L. tropica 208/48, L. tropica 101/65, L. guyanensis, L. braziliensis, L. major 159/06, L. major 230/119) were detected by primer set 1 (Fig. 3). The DNA from other protozoa (T. evansi, T. brucei, C. parvum, G. lamblia, E. histolytica) was not amplified, ensuring that differential diagnosis could be made with these primers.

Fig. 3. Specificity of the LAMP assay using primer set 1 on different Leishmania isolates at 65°C for 85 min. Lane 1, Leishmania major 159/06; 2, L. major 230/119; 3, L. infantum; 4, L. tropica 208/48; 5, L. tropica 101/65; 6, L. donovani THAK35; 7, L. guyanensis; 8, L. braziliensis; 9, Trypanosoma evansi; 10, negative control (distilled water); M, 100 bp DNA ladder.
The sensitivity of the primers was estimated with serially diluted DNA samples from various Leishmania spp. Under the conditions of 65°C for 85 min, the detection limit was variable for each Leishmania strain, ranging from 30 pg to 3·6 fg. The detection limits are shown in Table 2. The highest sensitivity was for the sample of L. tropica 101/65, for which the detection limit was found to be 3·6 fg, followed by L. major 230/119 at 12·6 fg and L. infantum L4 at 46 fg. The sensitivity for the sample of L. donovani THAK35 was determined to be the lowest at 30 pg, followed by the samples of L. major and L. tropica.
Table 2. Sensitivity of LAMP primer set 1 using different Leishmania strains under optimum amplification for set 1 (65°C for 85 min) and for set 2 (63°C for 85 min)

n.d., Not done
Testing LAMP primer set 2
The DNA of the same Leishmania isolates and heterologous DNA from other protozoan parasites were also tested using primer set 2 to determine the specificity of the primer set. DNA from all Leishmania isolates was amplified, whereas the DNA of the other parasites failed to provide a positive result. Therefore, primer set 2 is also specific for the detection of Leishmania spp.
Similar to the previous trials with primer set 1, after examining different amplification times and temperatures using all 10 strains, the optimal conditions for amplification were found to be 63°C and 85 min. To examine the sensitivity of LAMP primer set 2, the lowest concentrations of the amplified strain of each subtype of Leishmania that grew best during the previous trials were tested, namely L. donovani THAK35, L. infantum L4, L. major LV39 and L. tropica 101/65 (Fig. 4) at 63°C for 85 min. The results, shown in Table 2, indicate that primer set 2 is sensitive for the Leishmania spp. tested. The sensitivity for L. donovani THAK35 was 30 pg, and the sensitivity of L. infantum L4 and L. tropica 101/65 was 46·2 pg and to 3·6 pg, respectively. The only sample that was detected with greater sensitivity using this primer set was L. major LV39, with a detection limit of 1·6 fg.

Fig. 4. Sensitivity of the LAMP assay using primer set 2 on serially diluted DNA of Leishmania tropica 101/65 at 63°C for 85 min. Lane 1, 3·6 ng; 2, 360 pg; 3, 36 pg; 4, 3·6 pg; 5, 360 fg; 6, 36 fg; 7, 3·6 fg; 8, negative control; 9, positive control (L. infantum); M, 100 bp DNA ladder.
Results for PCR
PCR was performed, and the size of the PCR product was found to be 300–350 bp. This product represents the amplified part of the ribosomal ITS-1 DNA fragment. Using PCR, we successfully amplified DNA extracted from 1 × 105/5 μl to 1 × 101 parasites/5 μl of L. major DNA template. The results were visualized by gel electrophoresis and are shown in Figure 5. A DNA band of 300–350 bp can be seen in the first five lanes, whereas there is no amplification seen in lanes 6 and 7, which represent one parasite and the negative control, respectively. Considering that 1 fg is equivalent to ∼0·1 parasite [Reference Mary19], the detection limit of nested PCR was ∼100 fg.
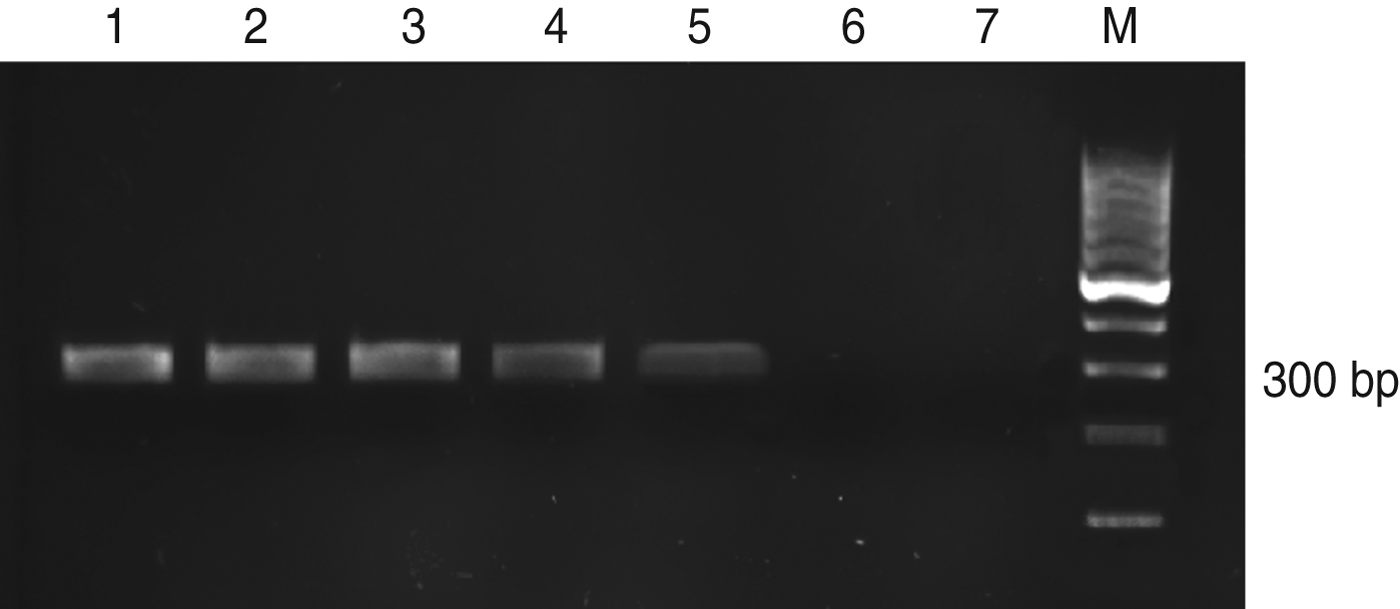
Fig. 5. PCR assay on serially diluted Leishmania major promastigotes. Lane 1, 105 promastigotes; 2, 104 promastigotes; 3, 103 promastigotes; 4, 102 promastigotes; 5, 10 promastigotes; 6, 1 promastigote; 7, negative control; M, 100 bp DNA ladder.
All of the examined DNA samples from different Leishmania spp. were successfully amplified by PCR assay. The DNA of the other tested organisms was not amplified, indicating that this assay was specific only for species of the Leishmania genus.
DISCUSSION
LAMP is a novel molecular method that may be used as an alternative method for the diagnosis of leishmaniasis [Reference Notomi20]. We developed and evaluated LAMP assays with two primer sets for the detection of a wide range of Leishmania spp. using DNA extracted from cultivated Leishmania promastigotes. Both primer sets were specifically designed to detect Leishmania spp. using only one test. These results indicate that different samples of L. major and L. tropica have variable sensitivity. However, it can be concluded that these primers are specific only for species of the Leishmania genus because they produced no false positive results for heterologous species in the LAMP assay. The DNA of 10 Leishmania spp. was amplified by both LAMP assays, whereas that of other parasites, including T. evansi and T. brucei, was not, indicating the specificity of both primer sets for the Leishmania genus. The control of the reaction with Trypanosoma DNA is of great importance because of the phylogenetic similarity between Leishmania spp. and Trypanosoma spp.
For primer set 1, the detection limit was different for each species, ranging from 30 pg to 3·6 fg. The Leishmania parasitaemia threshold is in the range of 1–8 parasites/ml [Reference Mary19]. The sensitivity was at the fg level, except for L. donovani THAK35. A possible explanation could be the presence of genetic heterogeneity in the subtypes. Primer set 2 also exhibited high sensitivity. However, its sensitivity was lower than that of primer set 1, with the exception of L. major LV39, for which the detection limit was 1·6 fg (a 100-fold increased sensitivity compared to primer set 1). In DNA samples containing L. infantum L4 and L. tropica, LAMP primer set 2 exhibited a 100-fold decrease in sensitivity compared to primer set 1. For the isolate L. donovani THAK35, the results were identical using both primer sets when an equal amount of DNA was amplified. Therefore, this primer pair could also be suitable for the detection of Leishmania DNA. The detection limit of the PCR assay was 1 × 101 parasites of L. major. As mentioned above, considering that 1 fg is equivalent to ∼0·1 parasite [Reference Grimaldi and Tesh21], the detection limit of PCR was ∼100 fg. Therefore, LAMP primer set 1 was more efficient than primer set 2.
The high sensitivity and specificity of LAMP primer set 1 indicates that it is an effective method for the successful detection of all pathogenic species of Leishmania. However, further trials must be undertaken, as our trials were performed with DNA from cultured promastigotes from the in vitro system. An evaluation with tissue samples is necessary so that this technology can be used to diagnose human and animal leishmaniasis, as LAMP-based point-of-care diagnostics should support the diagnosis of clinical leishmaniasis cases.
A LAMP assay for the amplification of L. donovani has been reported by Takagi et al. [Reference Takagi22] with a detection limit of 1 fg. The sensitivity appears to be high, but this assay was tested only with one Leishmania spp. In another study, a Leishmania assay was developed for the detection of several species in one test [Reference Adams23]. The sensitivity was between 10 and 100 parasites/ml, using an RT–LAMP assay with loop primers [Reference Adams23]. A disadvantage of the pan-RT–LAMP assay was the observation of false positives from T. brucei and T. cruzi due to the similarity of the 18S region between members of the Kinetoplastida class. The authors tried to underestimate the degree of cross-reactivity based on the different symptomatology of Chagas disease and leishmaniasis. They recommended the design of species-specific LAMP reactions or degenerate primer sets [Reference Adams23].
In the present paper, we describe a protocol for LAMP that is capable of amplifying a wide range of Leishmania spp. in a single test. PCR has been found to be a very good diagnostic method, and many studies of leishmaniasis are based on PCR. However, LAMP only requires basic equipment (no more than a water bath, when first evaluated and standardized) as the temperature is constant; it is therefore easier to perform in countries where leishmaniasis is endemic and costs must be minimized [Reference Adams23]. LAMP is a cheap test as any developed test has its own costs. Once the method is considered as developed and evaluated, the cost for the application of the method is significantly lower. We compared LAMP with a well optimized and sensitive PCR protocol which has been previously published [Reference Dweik18]. Large hospitals that possess a real-time turbidimeter would have the benefit not only of a fast and sensitive diagnosis in complicated or subclinical cases but also the opportunity to use LAMP as a quantitative method. In addition, a turbidimeter could be used for the diagnosis of a wide range of diseases. In the future, we expect that LAMP assays will accelerate investigations for the detection of Leishmania infections in humans and reservoir animals, including epidemiological/entomological investigations in sand flies, as material from infected sand flies can be used for LAMP assays. Further research is required for a definite assessment of the application of LAMP as a supplement or routine diagnostic method for the detection of Leishmania infection in hosts and vectors [Reference Kato24].
In conclusion, a new, rapid, simple and sensitive LAMP method for the diagnosis of Leishmania infection has been developed. We anticipate that this unified LAMP assay will contribute significantly to the diagnosis of the disease and can improve the convenient treatment and epidemiological studies of leishmaniasis in any setting worldwide, even if it does not differentiate between species of Leishmania. It is expected that this protocol will find applications in various countries using material from different sources and confirm the current assay as a useful and effective tool in the routine diagnosis of leishmaniasis.
ACKNOWLEDGEMENTS
We thank Dr Eleni Dotsika (Pasteur Institute Athens, Greece), Dr Patrick Scheid (Institute Medical Parasitology, Koblenz, Germany) and Dr Gabriele Schönian (Institute for Mikrobiologie und Hygiene, Charité Berlin, Germany) for supporting this study by supplying us with Leishmania parasites and DNA.
DECLARATION OF INTEREST
None.







