Sjögren’s syndrome is a chronic autoimmune disease characterized by dry eyes and dry mouth. In addition, a number of other organ systems with exocrine glands may be involved, and whole-body sicca syndrome may result. As recently as 2019, optic neuritis was reported as the initial manifestation of Sjögren’s syndrome. Reference Phuljhele, Pujari, Obedulla, Saxena and Sharma1 However, there have been increasing reports that the neurologic manifestations in Sjögren’s disease are better accounted for by concurrent diseases like neuromyelitis optica spectrum disease (NMOSD), specifically that associated with aquaporin-4 (AQP4)-IgG antibodies. Myelin oligodendrocyte glycoprotein (MOG)-IgG has more recently been recognized as a marker for patients with NMOSD seronegative for AQP4-IgG and in those with isolated or recurrent optic neuritis. We report a case of a patient who had Sjogren’s syndrome diagnosed after an initial attack of bilateral optic neuritis that was rediagnosed as MOG-IgG optic neuritis after a second recurrence and newly available testing.
A 53-year-old woman was referred for right eye reduced vision and pain with eye movements. She had a history of bilateral optic neuritis in both eyes 8 years prior to presentation that caused light perception in both eyes and was successfully treated with intravenous and oral corticosteroid treatment. As part of the workup for the initial optic neuritis, she was found to have positive anti-nuclear antibodies, positive Sjögren’s Syndrome-A extractable nuclear antibody, and positive rheumatoid factor. There were no clinical or radiological findings to support a diagnosis of multiple sclerosis as magnetic resonance imaging (MRI) of the brain was normal and lumbar puncture did not reveal cerebrospinal fluid (CSF)-specific oligoclonal bands. She also had objective signs of dry eye (Schirmer’s test supportive of aqueous tear deficiency and punctate epithelial staining on slit lamp examination) and complaints of dry mouth and arthralgias, further supporting the diagnosis of Sjögren’s syndrome. She was treated with hydroxychloroquine 200 mg daily. Her past medical history also included glomerulonephritis, hypothyroidism, and migraines. Her other medications included levothyroxine and candesartan.
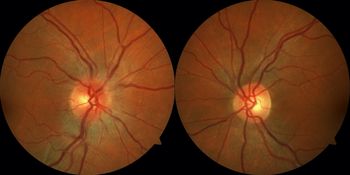
Eight years later, the patient represented with a 2-day history of right eye blurred vision and pain with eye movements. Initial ophthalmological examination revealed a visual acuity of 20/50 right eye and 20/20 left eye, a right relative afferent pupillary defect, and mild right optic disc edema (Figure 1). MRI of the brain and orbits revealed mild right optic nerve enhancement without significant perioptic enhancement with a normal-appearing brain. MRI of the spine was normal. Lumbar puncture revealed a normal protein and glucose level with one nonerythroid cell and no CSF-specific oligoclonal bands. She was treated with intravenous methylprednisolone 1 g daily for 3 days and had improvement in her vision over the next 2 weeks. Investigations for her optic neuritis revealed a positive serum MOG-IgG antibody (medium positivity) and negative aquaporin-4 antibody. Follow-up at 3 months revealed a visual acuity of 20/20 in both eyes and full Humphrey visual fields. No recurrence of the optic neuritis occurred at 6 months.

Figure 1: Fundus photographs demonstrate mild right optic disc edema while the left optic disc appears normal.
Neurologic manifestations have been observed in 10–60% of Sjögren’s syndrome patients, but these studies vary greatly in their inclusion criteria of symptomatic versus subclinical symptoms and diagnostic criteria used. Reference Chai and Logigian2 They are often grouped into peripheral and central manifestations, such as aseptic meningitis, transverse myelitis, brain and spinal cord lesions, and optic neuropathy, and small fiber neuropathy and sensory ataxic neuropathy, mononeuritis multiplex, polyradiculopathy, sensorimotor neuropathy, and autonomic neuropathy, respectively. Optic neuritis has been reported in 6–16% of patients with primary Sjögren’s syndrome in two retrospective cohort studies, but the literature remains sparse. Reference Chai and Logigian2 In addition, a literature review of optic neuritis in Sjögren’s syndrome shows that many patients are still not investigated for anti-AQP4 antibodies and apart from one case were not investigated for anti-MOG antibodies even as recently as a publication in 2019. Reference Phuljhele, Pujari, Obedulla, Saxena and Sharma1 Approximately 10–20% of patients with NMOSD are seronegative for AQP4. Reference Jarius, Franciotta and Paul3 Patients with Sjögren’s syndrome who experience optic neuritis but test negative for anti-AQP4 antibodies may be positive for anti-MOG antibody, given that these antibodies were only discovered and validated within the past decade. Primary Sjögren’s syndrome is the most common overlapping systemic autoimmune disorder with NMOSD. Reference Shahmohammadi, Doosti and Shahmohammadi4 As well, salivary gland biopsies from patients with NMOSD often reveal lymphocytic infiltration, which has been suggested to be due to the similar protein conformation of AQP4 in neuronal foot processes and AQP5 in salivary glands. Reference Javed, Balabanov and Arnason5 A similar link is yet to be revealed between MOG-IgG-associated diseases and Sjögren’s syndrome.
However, there is one previous report of MOG-IgG optic neuritis in a patient with Sjögren’s syndrome who reported xerophthalmia, xerostomia, and fatigue during the initial presentation of unilateral optic neuritis. Both conditions were diagnosed at the same time unlike our case where the patient carried a diagnosis of optic neuritis related to Sjögren’s syndrome. Reference Mittal, Baig and Merchant6 In addition, large cohort studies have shown patients with MOG-IgG-associated disease may have various associated autoimmune conditions, such as hypothyroidism, psoriasis, sarcoidosis, and Sjogren’s syndrome. Reference Cobo-Calvo, Ruiz and Maillart7 These associations hint these diseases may be closely related in clusters, though more research is needed to find common pathophysiological mechanisms. Given the increasing understanding that neurologic manifestations in a patient with Sjögren’s syndrome may be due to demyelinating syndromes, as seen in this patient with MOG-IgG optic neuritis, these symptoms should be treated with suspicion. A patient with Sjogren’s syndrome who experiences an episode of optic neuritis should be tested for anti-AQP4 and anti-MOG antibodies. The titre of these antibodies likely subsides after the acute attack and may not be detected during remission or following treatment with steroids or immunosuppressive agents. Patients with MOG-IgG often have good visual recovery following treatment and have a more optimistic prognosis, so an accurate diagnosis plays a pivotal role in improving patient outcomes.
In conclusion, our patient had coexisting diseases, Sjogren’s syndrome and anti-MOG-associated optic neuritis. Her initial optic neuritis resulted in a diagnosis of Sjogren’s syndrome, and she indeed had clinical and laboratory evidence for this condition. However, her optic neuritis was better accounted for by MOG-IgG-associated optic neuritis, and testing for this was not available at her initial presentation 8 years prior. We suggest that patients with a history of Sjogren’s syndrome and new optic neuritis are investigated with anti-AQP4 and MOG antibodies.
Disclosures
All authors have no disclosures to declare.