



Introduction
Neurodegeneration with brain iron accumulation (NBIA) belongs to neurodegenerative disorders. Its main feature is the accumulation of iron in several parts of the brain, especially in Globus Pallidus (GP). Reference Schneider1 The clinical features of NBIA include dystonia, parkinsonism, spasticity, cognitive impairment, and intellectual disability. Reference Di Meo and Tiranti2,Reference Schneider3 The age at onset varies from early childhood to late adulthood. Reference Schneider1 There is a lack of trustworthy information about the prevalence of NBIA in different geographical areas. The overall frequency of each subtype is shown in Figure 1. In fact, some areas such as the Dominican Republic are reported to have more cases of some NBIA subtypes. However, estimations are about 1/500,000 in some populations. There are different subgroups of NBIA of which pantothenate kinase-associated neurodegeneration (PKAN), phospholipase A2-associated neurodegeneration (PLAN), mitochondrial protein-associated neurodegeneration (MPAN), and Beta propeller-associated neurodegeneration (BPAN) are the most common forms. Mutations in pantothenate kinase 2 (PANK2), Phospholipase A2 Group VI (PLA2G6), Chromosome 19 Open Reading Frame 12 (C19orf12), and WD Repeat Domain 45 (WDR45) genes cause the aforementioned subgroups, respectively. Reference Hayflick, Kurian and Hogarth4 Proteins encoded by these genes mediate different cellular functions, including lipid metabolism, autophagy, coenzyme-A (CoA) biosynthesis, and mitochondrial and lysosomal function regulation. Reference Schneider1,Reference Di Meo and Tiranti2 NBIA inheritance patterns are autosomal recessive (AR), autosomal dominant, and X-linked. Reference Wiethoff and Houlden5 Diagnosis depends on clinical and magnetic resonance imaging (MRI) findings. In addition to color changes in MRI resulting from iron accumulation, some types of NBIA show a unique pattern in the MRI. The “eye of tiger” sign is the most well-known one in many cases of PKAN. Reference Sethi, Adams, Loring and El Gammal6 Since a significant number of patients remain without a proper diagnosis, genetic testing based on clinical features and MRI findings is beneficial. Reference Wiethoff and Houlden5 NBIA is classified based on disease-causing genes, which will be demonstrated later on.

Figure 1: The frequency of neurodegeneration with brain iron accumulation (NBIA) subtypes in the literature.
NBIA Classifications
Pantothenate Kinase-Associated Neurodegeneration; PKAN-NBIA1 (PANK2; Online Mendelian Inheritance in Man (OMIM)*606157)
PKAN was first described by two German physicians, Hallervorden and Spatz, so it was named Hallervorden–Spatz disease. However, due to their cooperation with the Nazi party today it is known as PKAN. It is the most common form of NBIA, which includes almost half of the cases (Figure 1). The exact prevalence of PKAN is yet unknown, but it is estimated to be 1–3/1,000,000. Reference Di Meo and Tiranti2,Reference Hayflick, Kurian and Hogarth4 It can occur from early childhood to middle age. There is an association between the age at onset and the disease course. The early-onset form of PKAN has a higher rate of progression than the late-onset form of the disease. Reference Hayflick, Kurian and Hogarth4
Although this NBIA is reported in many ethnicities and the dominant clinical picture is similar, there have been some variations in different populations. Lee et al. have found that cranial dystonias were less common in Asians than in Caucasians, and Asians tended to have segmental dystonia, whereas Caucasians were more likely to present generalized dystonia. In classic PKAN, dysarthria was more frequent in Asians, but parkinsonism in Caucasians. Pyramidal signs and mental impairment were less prevalent in Asians. Reference Lee, Lu and Chuang7
Classic Form of PKAN
This form is characterized by an early start and rapid progression. It occurs due to complete loss of function mutations in the PANK2 gene and often develops before the age of 6 years (average age at onset is 3 years old with a median survival of 9.5 years after onset). The main presentation of PKAN is considered to be a gait disturbance. Most of the affected children to the classic form of PKAN show a history of mild growth and developmental delay as well as dyspraxia before dystonia and spasticity. Pigmented retinopathy is a common feature in classical PKAN, but seizures are not. Although PKAN is known as neuroacanthocytosis disorder, the acanthocytosis, abnormal red blood cell morphology, is found in only some patients; since the high sensitivity genetic tests are available in many countries, this feature of erythrocytes is not a common diagnostic criterion. Reference Hayflick, Kurian and Hogarth4,Reference Chang, Zhang, Jiang, Wang and Wu8
Atypical Form of PKAN
In this form of PKAN, the clinical features are different from the classic form and start at later age (with a median age at onset of 18 years and 2% mortality in the 10 years follow up after onset). Reference Chang, Zhang, Jiang, Wang and Wu8 It is often presented with speech abnormalities. Progression happens at a slower pace, and it is the result of an incomplete loss of function mutations in the PANK2 gene. Other features of this form include dystonia and dysarthria. Among neuropsychiatric impairments in patients with the atypical form of PKAN, impulsivity and obsessive-compulsive disorder are the most common manifestations. Reference Hayflick, Kurian and Hogarth4 Pigmentary retinopathy is rare in this form, though extraocular movement abnormalities are common. Reference Hogarth9
Genetics of PKAN
The pattern of inheritance is AR and the disease results from mutations in the PANK2 gene encoding for pantothenate kinase 2 within the 20p13 chromosomal position. Reference Di Meo and Tiranti2,Reference Hartig, Hörtnagel and Garavaglia10 Currently, according to Human Gene Mutation Database (HGMD) professional 2019.4, 188 mutations in PANK2 have been reported. Chang et al. have proposed that the proportion of the patients carrying two null alleles is significantly higher in early-onset (classic) patients compared to late-onset (atypical) cases (27.5% vs. 0.8%). The null allele was presumed as “loss-of-function” allele. Reference Chang, Zhang, Jiang, Wang and Wu8
Pathophysiology of PKAN
PANK2 enzyme is a CoA sensitive sensor in the mitochondrial inner membrane. Reference Leonardi, Rock, Jackowski and Zhang11 PANK2 mutations lead to CoA metabolism impairments as a result of defects in the pantothenate kinase enzyme, leading to the accumulation of some of the CoA pathway substrates such as cysteine and compounds containing cysteine in basal ganglia. The result of these events is the iron accumulation in GP under the influence of rapid, toxic, and spontaneous cysteine oxidation, which leads to free radical production. Since basal ganglia and retina are sensitive to oxidative stress, the involvements of these sites are more prominent in PKAN patients. Reference Gregory and Hayflick12,Reference Bokhari and Bokhari13 A specific pattern of iron accumulation is detected in many PKAN cases, which is called the “eye of tiger” sign (Figure 2A and B). Reference Sethi, Adams, Loring and El Gammal6
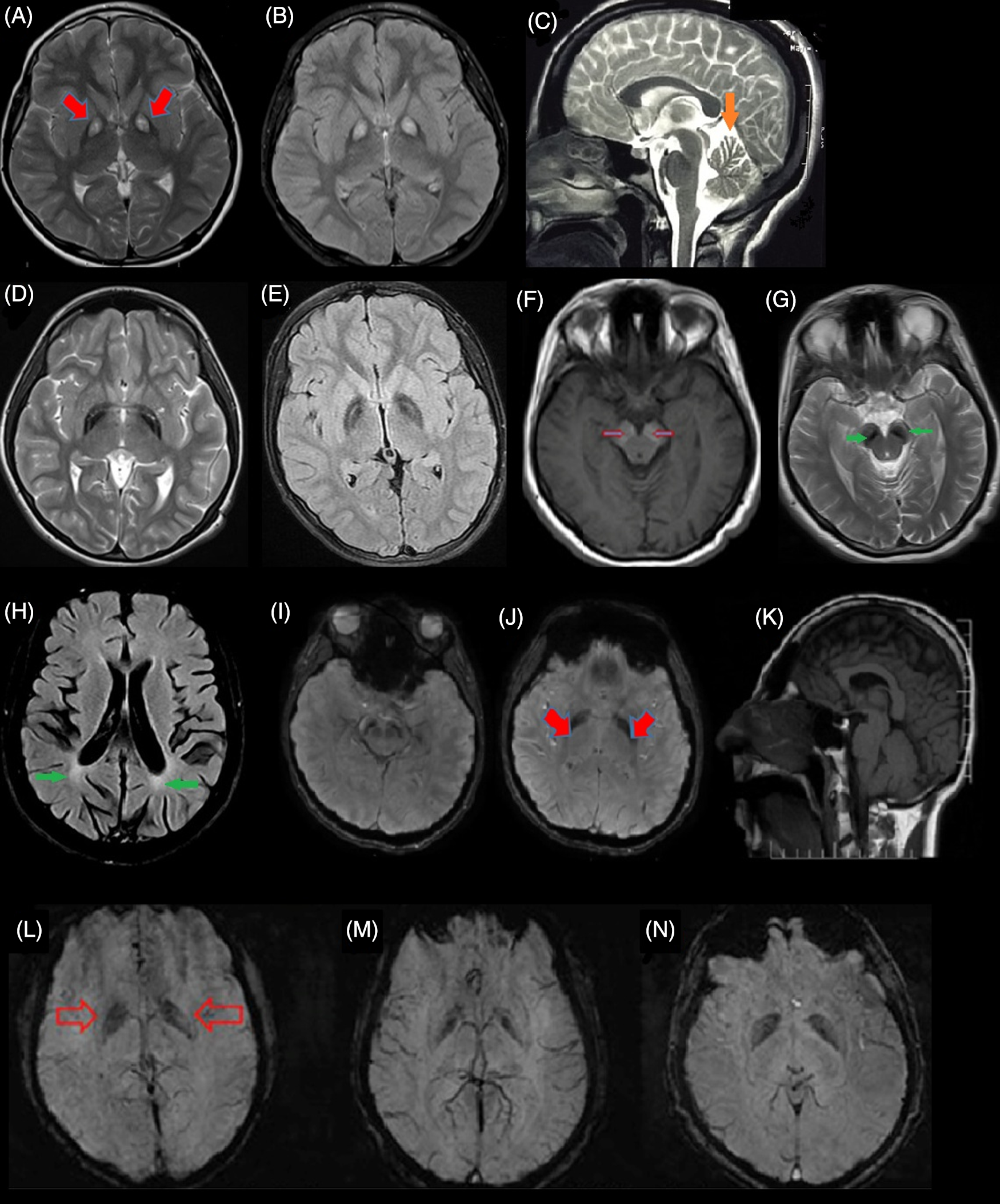
Figure 2: Brain magnetic resonance imaging (MRI) of different subtypes of NBIA from Iran. (A & B) Eye of tiger sign pattern (red arrow) in MRI of pantothenate kinase-associated neurodegeneration (PKAN) patients. (C) cerebellar atrophy (orange arrow) in phospholipase A2-associated neurodegeneration (PLAN). (D & E) iron deposition in globus pallidus (GP) with preserved medial medullary lamina in MPAN. (F & G) Halo sign in cerebral peduncles (substantia nigra [SN]) on T1-weighted sequences (empty red arrows) and iron deposition in the corresponding T2-weighted sequences in SN (green arrow) in a Beta propeller-associated neurodegeneration (BPAN) patients. (H) deep white-matter changes suggesting leukodystrophy (green arrows) in a fatty acid hydroxylase-associated neurodegeneration (FAHN) patient. (I–K) T2-weighted hypointensity signal in GP (red arrows) and SN and small pituitary gland (K) in the Woodhouse–Sakati syndrome (WSS) patients. (L–N) Susceptibility weighted imaging (SWI) in three affected to JABELS showing mild hypointensity in GP (empty red arrows) compatible with iron deposition.
Phospholipase A2-Associated Neurodegeneration; PLAN-NBIA2 (PLA2G6; OMIM*603604)
PLAN-NBIA2 comprises about 20% of NBIA cases (Figure 1), manifesting a broad range of symptoms, from psychological disorders in childhood to parkinsonism–dystonia in adulthood. Pathological abnormalities in the central nervous system (CNS) and peripheral nervous system (PNS) have been detected. Reference Di Meo and Tiranti2,Reference Hayflick, Kurian and Hogarth4 This type of disease is categorized into three phenotypic forms with clinical and radiological overlap.
Infantile-Onset PLAN or Infantile Neuro-Axonal Dystrophy (INAD)
Early-onset form of disease emerges between the age of 6 months to 3 years (around 2 years) with growth retardation, hypotonia, and spastic tetraparesis. Reference Gregory, Westaway and Holm14–Reference Guo, Tang and Guo16 Many of the affected children can never walk. Seizure and ophthalmological impairment (strabismus, nystagmus, and optic atrophy) are prominent in these patients and death occur in infancy and early childhood. Reference Guo, Tang and Guo16
Childhood-Onset PLAN or Atypical Neuro-Axonal Dystrophy
Atypical neuro-axonal dystrophy shows more phenotypic variations in comparison with INAD. The age at onset can vary from early childhood (usually after 3 years) to the end of the second decade of life. These children show ataxia, dystonia, spasticity, delay in speaking, and autistic symptoms with degrees of intellectual disability. Strabismus, nystagmus, optic atrophy, and neuropsychological disorders are also common. These patients have slower progression and longer survival times compared to INAD cases. Reference Hayflick, Kurian and Hogarth4,Reference Guo, Tang and Guo16
Adult-Onset PLAN or Dystonia–Parkinsonism
Patients affected with this form present their symptoms at various ages, but most patients present symptoms early in adulthood (20–40 years). Patients manifest dystonia and Parkinsonism from late adolescence to early twenties and it might be accompanied by mild mental impairment. Survival for several decades is not unusual. Reference Hayflick, Kurian and Hogarth4,Reference Guo, Tang and Guo16,Reference Paisan-Ruiz, Bhatia and Li17
Genetics of PLAN
Mutation in PLA2G6, encoding for phospholipase A2, within 22q13.1 chromosomal position results in PLAN. The pattern of inheritance is considered AR. Reference Morgan, Westaway and Morton18 Most of the patients have missense mutations that do not disrupt the catalytic activity of the protein. Reference Engel, Jing, O’Brien, Sun and Kotzbauer19 . Currently, according to the HGMD professional 2019.4, 191 mutations in PLA2G6 have been reported.
Pathophysiology of PLAN
PLA2G6 encodes for a specific type of phospholipase, which catalyzes the release of fatty acid from phospholipid. It has been proposed that this gene plays a crucial role in phospholipid homeostasis, and the impairment of this enzyme increases the level of phospholipid and acetyl-CoA. Impairment in phospholipid homeostasis may change the lipid components of membrane, vesicles, or endosomes, resulting in membrane permeability, fluidity, iron homeostasis disturbance, and disruption of the autophagy process. Reference Malik, Turk and Mancuso20,Reference Shinzawa, Sumi and Ikawa21 Alpha-synuclein and neurofibrillary tangles in the pathologic study of PLAN have suggested a link to Parkinson’s disease. Although some studies have implicated that PLA2G6 mutation is not a major risk factor for Parkinson’s disease in Asian populations. Reference Guo, Tang and Guo16
Mitochondrial Protein-Associated Neurodegeneration; MPAN-NBIA4 (C19orf12; OMIM*614297)
Historically, this type of NBIA was first discovered in a group of patients in eastern Europe. Reference Hartig, Iuso and Haack22 Global prevalence is estimated to be fewer than 1/1,000,000 and until now less than 100 patients are confirmed to be affected to MPAN. The clinical features include dystonia, Parkinsonism, spasticity, and psychological problems such as anxiety, depression, inattention, hallucination, and hyperactivity. Onset is usually in the first or second decade (median onset of 9 years), but adult-onset cases are also reported. Reference Sparber, Krylova and Repina23 The late-onset form of disease starts in the third or fourth decade of life with disorders like psychosis and dementia. Reference Hayflick, Kurian and Hogarth4,Reference Hartig, Iuso and Haack22,Reference Hogarth, Gregory and Kruer24 The disease severity and progression of MPAN are thought to be generally similar or milder than PKAN but data on this are still lacking due to the rarity of this NBIA and the absence of large cohorts. There are reports of cases with preserved ambulation after two decades of symptom onset. Reference Hartig, Iuso and Haack22–Reference Schulte, Claussen and Jochim25
Genetics of MPAN
MPAN is caused by mutations in C19orf12 on 19q12, a gene encoding a mitochondrial membrane protein. Reference Hartig, Iuso and Haack22 The pattern of inheritance is AR, but there are reports of autosomal dominant in some cases. Reference Hogarth, Gregory and Kruer24 Currently, according to the HGMD professional 2019.4, 51 mutations in C19orf12 have been reported. The most frequent type of mutations affecting this gene are p.Gly69Argfs*10 and p.Thr11Met. Reference Schneider3 Landoure and colleagues reported c.187G>C; p.Ala63Pro mutation in two sisters affected to hereditary spastic paraplegia type-43 (HSP-SPG43) without iron accumulation in their brain MRI. This mutation was also reported in at least two other families affected by MPAN, thus the determination of genotype–phenotype correlation is challenging. Reference Gregory, Hartig, Prokisch, Kmiec, Hogarth and Hayflick26
MPAN Pathophysiology
C19orf12 encodes for a small transmembrane protein localized in the mitochondrial membrane. The precise functionality remains to be unraveled, but based on several studies, fatty acid biosynthesis and destruction of branched amino acids (valine, leucine, and isoleucine) are two prominently involved pathways. Reference Hartig, Iuso and Haack22 Studies on postmortem affected samples showed increased alpha-synuclein in stained tissue and Lewy body formation in the brain. Also, it seems that spheroidal eosinophilic structures in GP are byproducts of neuronal degeneration. More investigations are needed to understand the exact pathophysiology of mutations in this gene. Reference Hogarth, Gregory and Kruer24
Beta Propeller-Associated Neurodegeneration; BPAN-NBIA5 (WDR45; OMIM*300526)
The initial phenotype includes neural developmental delay in early childhood, intellectual disability (autism or a Rett-like syndrome), seizures, and sleep problems. Reference Haack, Hogarth and Kruer27,Reference Hayflick, Kruer and Gregory28 Subsequently, speech skills become impaired. Ataxia, dystonia, and Parkinsonism are later features that add to the clinical picture in adolescence or early adulthood (at a mean age of 27 years). Reference Hayflick, Kurian and Hogarth4,Reference Stige, Gjerde, Houge, Knappskog and Tzoulis29 The initial non-motor symptoms may be mild or moderate and the clinical picture discloses itself by later motor symptoms and typical MRI features, which may be nonspecific or insignificant in early childhood imaging. Reference Chard, Appendino and Bello-Espinosa30,Reference Kimura, Sato and Ishiyama31
Genetics of BPAN
BPAN is the only form of NBIA with an X-linked dominant pattern of inheritance, resulting from mutations in the WDR45 gene on Xp11.23. This gene encodes an autophagy protein, beta-propeller. Reference Haack, Hogarth and Kruer27 Up to now, most of the BPAN cases were as a result of de novo mutations, and some affected men were mosaic; probably because of the post-zygotic mutations. By the development of next generation sequencing and whole exome sequencing (WES), many cases were identified and the spectrum of clinical features has been widening. Reference Hayflick, Kurian and Hogarth4 Currently, according to the HGMD professional 2019.4, 102 mutations in WDR45 have been reported.
Pathophysiology of BPAN
In this form of the disease, there are numerous findings indicating autophagy pathway impairments. WDR45 protein is a member of a protein family that facilitates the formation of the protein complex and plays an essential role in many biological processes such as signaling, apoptosis, and gene expression regulation. WDR45 protein can bind to Autophagy Related 2A (ATG2A) and Autophagy Related 2B (ATG2B) that are two other autophagy-related proteins, indicating the affiliation of this protein in the autophagy process. Reference Behrends, Sowa, Gygi and Harper32,Reference Meyer, Kurian and Hayflick33 Lymphoblastic cell line in patients affected to BPAN, with severe decrease of WDR45 protein and lower autophagy function rate leads to abnormal accumulation of initial autophagy structures. Reference Saitsu, Nishimura and Muramatsu34 Neuropathological investigations of BPAN affected patients showed severe neurodegeneration, axonal spheroids, siderophages, and reactive astrocytes. Abnormality in the autophagy process leads to the accumulation of degraded components in the cell, toxic conditions, and cell stress resulting in neuronal degeneration. Reference Hayflick35 Iron accumulation in BPAN patients in substantia nigra (SN) resembles a halo sign pattern on T1-weighted sequences of MRI Reference Kruer, Hiken and Gregory36 (Figure 2F).
Coasy Protein-Associated Neurodegeneration; CoPAN-NBIA6 (Coenzyme A synthetase (COASY); OMIM*609855)
CoPAN-NBIA is a rare form of NBIA reported in 2014 in two Italian patients. Reference Dusi, Valletta and Haack37 Clinical features manifested in the first decade with mild cognitive impairment and gait disturbance. Then through the second decade, oromandibular dystonia, dysarthria, and progressive spasticity, and axonal neuropathy were pursued. The complete severe phenotype in the third decade included exacerbation of prior symptoms as well as the emergence of parkinsonism, depression, and obsessive-compulsive disorder. Reference Schneider1,Reference Hayflick, Kurian and Hogarth4,Reference Hogarth9,Reference Dusi, Valletta and Haack37–Reference Annesi, Gagliardi, Iannello, Quattrone, Iannello and Quattrone39
Genetics of CoPAN
Mutations in COASY, showing an AR pattern of inheritance, are the genetic causes of CoPAN. COASY is located on 17q21, which encodes the CoA-synthase enzyme. Reference Dusi, Valletta and Haack37 Currently, according to the HGMD professional 2019.4, five mutations in COASY have been reported.
Pathophysiology of CoPAN
COASY is a critical enzyme in CoA biosynthesis. Defects in this gene lead to a reduction of CoA and acetyl-CoA levels. Acetyl-CoA is involved in the regulation of the autophagy process. Investigation of CoPAN patients shows strong links between lipid metabolism, CoA synthesis, and autophagy in NBIA pathology. In patients’ fibroblasts culture, it has been shown that COASY protein and acetyl-CoA levels are lower than usual significantly. Reference Meyer, Kurian and Hayflick33,Reference Dusi, Valletta and Haack37,Reference Martinez, Tsuchiya and Gout40,Reference Mariño, Pietrocola and Eisenberg41
Fatty Acid Hydroxylase-associated Neurodegeneration; FAHN (FA2H; OMIM*611026)
Clinical hallmarks of this disease include ataxic spastic gait, starting in the first decade (median age of 4 years) followed by a decline in intellectual function, dystonia, seizure, divergent strabismus, and exacerbated cerebellar and pyramidal signs in the second decade. With the disease progression, as a matter of time (median 7 years), dystonia and spasticity limit the ability of movement; thus, patients require a wheelchair in the adolescence or early twenties. Reference Schneider1,Reference Hayflick, Kurian and Hogarth4,Reference Kruer, Paisán-Ruiz and Boddaert42–Reference Mari, Berti and Romano44
Genetics of FAHN
Defects in this gene were described in HSP (HSP-SPG35) and since then, mutations in this gene were introduced in movement disorders with brain iron accumulation. Reference Kruer, Paisán-Ruiz and Boddaert42 Nowadays, HSP-SPG35 is regarded as a type of FAHN disease because of the pattern of iron accumulation in basal ganglia in many reported cases. Reference Hayflick, Kurian and Hogarth4 FAHN is due to AR mutations in FA2H on 16q23.1, Reference Di Meo and Tiranti2 also there are some reports of uniparental disomy. Reference Gregory, Venkateswaran, Hayflick, Adam, Ardinger and Pagon45 Currently, according to the HGMD professional 2019.4, 65 mutations in FA2H have been reported.
Pathophysiology of FAHN
FA2H enzyme is a 2-hydroxylase enzyme expressed in Schwann cells and oligodendrocytes. Reference Alderson, Rembiesa, Walla, Bielawska, Bielawski and Hama46 2-hydroxy sphingolipids produced by FA2H are the most abundant lipids in the myelin sheath. Mutations in this gene lead to a decreased level of enzyme activity and, subsequently, myelin sheath degeneration. Reference Meyer, Kurian and Hayflick33,Reference Zöller, Meixner and Hartmann47 FA2H is also a lipid raft movement regulator. Lipid rafts are cholesterol and sphingolipid structures of membrane involved in membrane transporting and signaling. Guo and colleagues showed that FA2H-dependent hydroxylation affects membrane protein transport due to lysosomal degradation. Based on these findings, defects in FA2H lead to changes in the 2-hydroxy sphingolipid profile that can affect membrane lipids. Reference Meyer, Kurian and Hayflick33,Reference Guo, Zhou, Pryse, Okunade and Su48
Aceruloplasminemia (CP; OMIM*117700)
Aceruloplasminemia is one type of NBIA with direct relation to iron homeostasis and is reported globally, but it seems to be more common in the Japanese population (1/2,000,000). Accumulation of iron in the brain and other organs leads to neurodegeneration, retinopathy, and diabetes mellitus. Neurologic symptoms mainly include movement disorders including ataxia, chorea, dystonia, and tremor with cognitive and psychological changes. The age at onset is mainly in early to middle adulthood (median age of neurologic symptom onset, 51 years). Reference Hayflick, Kurian and Hogarth4,Reference Wiethoff and Houlden5,Reference Vroegindeweij, Boon, Wilson and Langendonk49 Diabetes is reported in almost 70% of affected individuals with microcytic or normocytic anemia because of iron accumulation in the pancreas. Since diabetes mellitus and microcytic anemia often occur one or two decades earlier than neurologic signs (median age, 35 years), it is an important clue in the early diagnosis of patients. Reference Vroegindeweij, Boon, Wilson and Langendonk49–Reference Vroegindeweij, Van Der Beek and Boon51 A mortality of 30% is reported in patients under chelation therapy after a median of 7 years of neurological symptoms onset (mean age at death, 60 years). Reference Vroegindeweij, Boon, Wilson and Langendonk49
Genetics of Aceruloplasminemia
Loss of function mutations in the CP gene results in the AR form of NBIAs, called aceruloplasminemia. CP is located on 3q21–24 and encodes a plasma membrane glycoprotein that plays a role in iron transport and processing in a way that helps the binding of iron to transferrin. Transferrin transports the iron to the red blood cells to carry oxygen. Mutations in CP lead to unstable and nonfunctional protein, which does not transport the iron out of cells and its accumulation will damage the cells and tissues. Currently, according to the HGMD professional 2019.4, 65 mutations in this gene have been reported. Reference Levi and Finazzi52,Reference McNeill, Pandolfo, Kuhn, Shang and Miyajima53
Pathophysiology of Aceruloplasminemia
Ceruloplasmin is the only ferroxidase, which is expressed by astrocytes. Defects in this protein lead to a disability of astrocytes in oxidizing the iron that is directly being transferred to CNS. Reference Brissot, Ropert, Le Lan and Loréal54 There are two forms of ceruloplasmin protein; the secretory form that is mainly expressed in the liver and the GPI-linked form that is mainly expressed in the brain. Reference Kono, Yoshida and Tomosugi55 Based on in vivo and in vitro studies, several factors contribute to the pathogenesis of disease such as: (1) ceruloplasmin dysfunction that leads to iron accumulation; (2) oxidative stress and the reactive oxygen species (ROS) production; (3) increased level of lipid peroxidation; and (4) change in lipid environment that leads to mitochondrial dysfunction and neuronal degeneration. Reference Miyajima, Takahashi and Kono56
Neuroferritinopathy-NBIA3 (FTL; OMIM*134790)
Neurological signs such as chorea, dystonia, cerebellar symptoms, psychosis, depression, cognitive impairment, Parkinsonism, dysarthria, insomnia, sleep apnea, areflexia, a positive Romberg test, and dysautonomia are the clinical features of this type of NBIA. Reference Chinnery, Crompton and Birchall57–Reference Kumar, Rizek and Jog59 The onset is between the second to fifth decades (mean age, 40 years) depending on the type of mutation. Neuroferritinopathy progresses gradually but in those with c.458dupA mutation a rapid progression of Parkinsonism, ataxia, and neuropsychiatric symptoms has been reported. Death is mostly due to aspiration pneumonia. Reference Kumar, Rizek and Jog59
Genetics of Neuroferritinopathy
Neuroferritinopathy is due to mutations in the FTL gene, which encodes the light chain of ferritin. Ferritin is regarded as the main iron storage protein. FTL is located on 19q13.33. Mutations in this gene are transmitted by an autosomal-dominant pattern of inheritance with complete penetrance. Most of these mutations cluster in the fourth exon. Cataract hyperferritinemia syndrome is due to a mutation in the 5′-untranslated region (5′UTR) in FTL with no neurological symptoms. Reference Di Meo and Tiranti2,Reference Hayflick, Kurian and Hogarth4 Currently, according to the HGMD professional 2019.4, 66 mutations in FTL have been reported, and interestingly, 36 mutations are in regulatory regions of the gene.
Pathophysiology of Neuroferritinopathy
The affected individuals’ fibroblast culture revealed the accumulation of ferritin in the cytoplasm and nucleus, the lower expression of transferrin receptor, and the higher expression of divalent metal transporter 1 and ROS formation. Defect in ferritin leads to a higher level of unstable iron in cells, therefore increases the ROS production and oxidized protein, decreasing the proteasome proficiency. Reference Cozzi, Rovelli and Frizzale60 Pathologic investigations showed ferritin inclusion body in the cytoplasm and nucleus of glia and CNS neurons and also in other organs related to iron.
Kufor-Rakeb Syndrome; KRS (ATPase Cation Transporting 13A2 (ATP13A2); OMIM*610513)
It is a rare type of atypical young onset Parkinsonian syndrome, which occurs before the age of 20 years (Parkinson disease 9 (PARK9)). “Kufor-Rakeb” is the name of a region in Jordan, where it was first reported. Reference Najim al-Din, Wriekat, Mubaidin, Dasouki and Hiari61 Clinical features like visual hallucinations, dementia, perioral myokymia (facial-faucial-finger mini myoclonus), supranuclear ophthalmoplegia, and spasticity have been reported. Since, in some cases, the iron accumulation in basal ganglia is detectable on brain MRI, it is suggested to be considered as NBIA syndrome. The Parkinsonism responds to levodopa but is complicated by the early development of motor fluctuations. Reference Hayflick, Kurian and Hogarth4,Reference Wiethoff and Houlden5,Reference Hogarth9,Reference Schneider, Paisan-Ruiz and Quinn62
Genetics of Kufor-Rakeb Syndrome
It is caused by mutations in ATP13A2 located on 1p36.13, encoding a p-type ATPase. Affected individuals show juvenile Parkinsonism, and in some cases, there are reports of iron accumulation in basal ganglia. Reference Kruer, Paudel and Wagoner63,Reference Ramonet, Podhajska and Stafa64 The pattern of inheritance is AR and currently, according to the HGMD professional 2019.4, 53 mutations in ATP13A2 have been reported. Mutations in this gene were also reported in autosomal recessive-hereditary spastic paraplegia (AR-HSP), spastic paraplegia 78 (SPG78). The disease emerges in adulthood, presenting mainly with spasticity and rarely Parkinsonism. Reference Estrada-Cuzcano, Martin and Chamova65
Pathophysiology of Kufor-Rakeb Syndrome
In vitro studies show that mutant ATP13A2 protein remains in the endoplasmic reticulum (ER), and then is degraded by the proteasome. The mutant protein accumulation can lead to ER stress induction. Reference Park, Mehta and Cooper66,Reference Ramirez, Heimbach and Grundemann67 Investigation of affected individual cultured fibroblast shows that lack of ATP13A2 leads to lysosomal dysfunction because of vesicle structures (lysosomes and autophagy vacuoles) accumulation and proteolytic dysfunction of the lysosomal enzymes, and disability to digest lysosomal substrates. Reference Dehay, Ramirez and Martinez-Vicente68,Reference Usenovic, Tresse, Mazzulli, Taylor and Krainc69 Deficiencies in the mitochondrial respiratory chain lead to a decrease in ATP production. In Kufor-Rakeb patients, mitochondrial DNA is fragmented, and its integrity is lost. Reference Grunewald, Arns and Seibler70
Woodhouse–Sakati Syndrome; WSS (DDB1 And CUL4 Associated Factor 17 (DCAF17); OMIM*612515)
This form of NBIA is a rare AR disorder, which occurs in adolescence. WSS consists of mixed neurologic and endocrine features including deafness, alopecia, progressive dystonia, dysarthria, dysphagia, cognitive impairment, intellectual disability, hypogonadism, and diabetes mellitus. Some patients show iron accumulation in GP and SN (Figure 2I–K). WSS occurs due to mutations in the DCAF17 gene on 2q31.1 that encodes a transmembrane nuclear protein with unknown function. Reference Alazami, Al-Saif and Al-Semari71,Reference Jin, Arias, Chen, Harper and Walter72 Currently, according to the HGMD professional 2019.4, 21 mutations in DCAF17 have been reported mainly in the Middle East. Two clinical subtypes based on the severity and progressive disabilities (mainly due to neurological symptoms) are proposed. The severe subtype manifests earlier (mean age, 12.6) with progressive generalized dystonia leading to disability in mean 7.4 years whereas the second type starts later (mean age, 18.1) with focal limb or cervical dystonia and patients remain ambulatory for at least two decades. Reference Bohlega, Abusrair and Al-Ajlan73
Jaberi–Elahi Syndrome; JABELS SYNDROME (GTPBP2; OMIM*607434)
This form is also known as neurodevelopmental retardation with a delay in growth and intellectual disability. It starts in childhood, and the severity of symptoms is variable. Some patients have social interactions, with ataxic gait, tremor, or dystonia, while others might not achieve any movement control and are not able to speak. Optic anomaly, microcephaly, abnormality in hand and foot structure, kyphoscoliosis, dysmorphic face, and seizures are their other features. MRI usually shows cerebellar and corpus callosum hypoplasia and atrophy. Recently, Jaberi and colleagues reported an Iranian family with consanguineous marriage in which four affected children with neurodevelopmental impairment and age at onset in childhood were born. All affected children showed psychomotor retardation including mild delay in motor milestones, speech delay, intellectual disability, and behavior abnormalities. MRI findings indicated cerebellum atrophy, hypointensity in GP, and SN indicating abnormal iron deposition (Figure 2L–N). Homozygosity mapping and WES analysis in this family revealed a homozygous splice-site variant, in the GTPBP2 gene. Reference Jaberi, Rohani and Shahidi74 GTPBP2 is located on 6p21.1 and encodes a GTP-binding protein, which participates in the reporter mRNA stability but the precise function is still unknown. Reference Woo, Kim and Lee75 GTP-binding protein 1 (GTPBP1) and GTPBP2 are members of the GTPase superfamily, which participate in cell proliferation, differentiation, and protein synthesis. Reference Bourne, Sanders and McCormick76,Reference Kaziro, Itoh, Kozasa, Nakafuku and Satoh77
Leukoencephalopathy with Dystonia and Motor Neuropathy; LKDMN (Sterol Carrier Protein 2 (SCP2); OMIM*184755)
LKDMN is an AR disorder, reported in 2006 for the first time in an adult person with dystonic head tremor, spasmodic torticollis, azoospermia, hypogonadotropic hypogonadism, mild cerebellar ataxia, gait impairment, and hyposmia. Plasma metabolic analysis revealed pristanic acid accumulation and abnormal excretion of bile alcohol glucuronides in the urine. Brain MRI revealed bilateral hyperintensity signal on thalami, Pons, and occipital lobes. Reference Ferdinandusse, Kostopoulos and Denis78 Molecular analysis revealed a homozygous missense mutation in SCP2. Recently, the second affected individual, a 51 years old man with a compound heterozygous missense mutation in this gene, was identified. He had movement impairment and ataxia with NBIA features in MRI but no leukoencephalopathy. Reference Horvath, Lewis-Smith and Douroudis79 The SCP2 gene is located on 1p32.3 and encodes two different proteins; sterol carrier protein X (SCPx) and SCP2 that are result of transcription initiation with two unrelated regulatory promoters. Reference Seedorf, Raabe and Ellinghaus80 Both these products are peroxisomal enzymes with thiolase activity utilized for branched-chain fatty acids breakage. Pathologic hallmarks in this disorder are probably due to the branched chain fatty acids accumulation. Reference Wanders, Vreken and Ferdinandusse81 Affected individuals with mutations in SCP2 may have an abnormality in fatty acid acetyl-CoA metabolism, which is a common mechanism in NBIA pathogenicity. This fact indicates that iron accumulation in the brain can be a secondary phenomenon due to metabolic defects. By this hypothesis, iron chelation-based therapeutics remain uncertain. Reference Horvath, Lewis-Smith and Douroudis79
New Subtypes of NBIA
NBIA7 (RalA Binding Protein 1 (RALBP1) Associated Eps Domain Containing 1 (REPS1); OMIM*614825)
Drecourt and colleagues reported two sisters with unrelated healthy parents who originated from France. The clinical features in the first sister were speech and motor delay, hypotonia, progressive cerebellar ataxia, losing the ability to walk at the age of nine, and nystagmus. Dysarthria, dysmetria, spasticity of the lower limbs, and pes cavus were also reported. Cerebellar and cerebral atrophy and iron accumulation in the GP and cerebral peduncles showed on the brain MRI. She eventually died at the age of 20 years. The second sister had the same but milder features. She can walk yet but with help at the age of 14 years. WES identified a compound heterozygous variant, c.232G>C and c.338C>A, in the REPS1 gene. Both variants were in an epsin-15 homology domain, a conserved region that interacts with RALBP1 to form the endosome recycling compartment. REPS1 is located on 6q24.1 and encodes RALBP1-associated Eps domain-containing protein-1. This protein is affiliated to autophagy because of its involvement in endocytosis, vesicle transport, localization in the cytoplasm, and endosome. This type of disease is known as NBIA7. Reference Drecourt, Babdor and Dussiot82,Reference Levi and Tiranti83
NBIA8 (Carnitine O-Acetyltransferase (CRAT); OMIM*600184)
Drecourt et al. also reported another subtype of NBIA in a Turkish girl with first cousin parents. Clinical manifestations include hypotonia, brisk deep tendon reflexes, sensory neuropathy, tremor, dysmetria with evidence of cerebellar atrophy, posterior leukoencephalopathy, hyperintensity of basal ganglia, hypointensity of GP, and SN which is suggestive for iron accumulation. Single-nucleotide polymorphism (SNP) genotyping and WES revealed a homozygous variant c.962G>A in the CRAT gene. This gene is located on 9q34.11 and encodes carnitine O-acetyltransferase. The encoded protein is localized in mitochondria and catalyzes the reversible transfer of acyl groups from carnitine to CoA and regulates the acyl-CoA/CoA ratio. It also plays a crucial role in the transport of fatty acids for beta-oxidation. This type of disease is known as NBIA8. Reference Drecourt, Babdor and Dussiot82,Reference Levi and Tiranti83
Hereditary Spastic Paraplegia-50; HSP-SPG50 (Adaptor Related Protein Complex 4 Subunit Mu 1 (AP4M1); OMIM*602296) or AP4M1-Associated NBIA
This gene encodes a subunit of the heterotetrameric AP-4 complex. AP4 is composed of four chains: beta-4 and epsilon-4 (AP4B1 and AP4E1 large chains), mu-4 (AP4M1 medium-chain), and sigma-4 (AP4S1 small chain). AP4M1 gene is located on 7q22.1 and encodes the mu subunit of the adaptor protein complex-4. This gene is ubiquitously expressed in the CNS and plays a role in vesicle formation, post-Golgi protein trafficking, and sorting processes. Reference Levi and Tiranti83,Reference Tüysüz, Bilguvar and Koçer84 Although mutations in genes encoding the different subunits of adaptor protein complex-4 have been reported in patients affected to different types of HSP, Roubertie and colleagues in 2018 reported the clinical features of three patients originating from a large consanguineous Moroccan family who exhibited early-onset developmental delay, juvenile motor function deterioration, spasticity, and pyramidal tract signs and intellectual deficiency in which brain MRI findings were compatible with disorders in the NBIA spectrum. Their MRI showed the isointense pattern of the GP and hypointensity of the GP on T1 and T2 sequences, respectively. WES was performed and revealed a homozygous mutation c.916C>T;p.Arg306* in the AP4M1 gene. These data show the overlap between NBIA and hereditary spastic paraplegia. Reference Roubertie, Hieu and Roux85 Previously, Najmabadi and colleagues reported a mutation in AP4M1 in a patient with microcephaly and paraplegia. Reference Najmabadi, Hu and Garshasbi86
PARK15 (FBXO7; OMIM*605648) or FBXO7-Associated NBIA
The F-box only protein 7 (FBXO7) gene is located on 22q12.3 and encodes a protein involved in the ubiquitin–proteasome protein-degradation pathway and mitophagy. In 2008, Shojaee and colleagues performed a linkage analysis in a large Iranian family with Parkinsonian-pyramidal syndrome and found a region on chromosome 22 linked to disease. Sequencing was done for candidate genes in that region which revealed a disease-associated variant in the FBXO7 gene, PARK15. Recently Correa-Vela and colleagues reported a novel homozygous mutation, c.368C>G; p.S123*, in FBXO7 in a child with spastic paraplegia, epilepsy, cognitive decline, cerebellar degeneration, levodopa nonresponsive Parkinsonism. Brain MRI revealed iron deposition in the GP and SN appeared as a hypointense signal on iron-sensitive sequences which is compatible with NBIA. All these findings suggest that FBXO7-associated NBIA can be a novel subtype of the NBIAs and there are clinically and genetically overlap between NBIA and other neurodegenerative disorders. Reference Correa-Vela, Lupo and Montpeyó87,Reference Shojaee, Sina and Banihosseini88
Differential Diagnosis of NBIA
Clinical features of NBIA are often diverse and involved different systems like pyramidal, extrapyramidal, cortical, and cerebellar, so the differential diagnosis consists of a broad spectrum of disorders such as inborn errors of metabolism, lysosome storage diseases, hereditary spastic paraplegia, and other forms of parkinsonism–dystonia. Reference Grunewald, Arns and Seibler70 T2-weighted hypointensity in GP is also reported in cases with mutations in Lysine Methyltransferase 2B (KMT2B) (KMT2B-related dystonia), Solute Carrier Family 39 Member 14 (SLC39A14) (SLC39A14-related early-onset dystonia–parkinsonism) and Sequestosome 1 (SQSTM1) (neurodegeneration with ataxia, dystonia, and gaze palsy, childhood-onset [NADGP]) (https://www.ncbi.nlm.nih.gov/books/NBK121988/). On the other hand, the lack of iron in the early stage of disease makes the diagnosis more challenging. It is noteworthy that the level of iron in the brain is deeply dependent on age in a way that there is no iron accumulation at birth but tends to increase during development. Reference Schneider3 Diagnostic clues for the main subtypes of NBIA are summarized in Figure 3.

Figure 3: Diagnostic clues for main subtypes of NBIA.
Molecular Mechanism of NBIA Proteins
Different cellular and molecular pathways are mediated by NBIA proteins such as CP and FTL, which are iron homeostasis regulator; PLA2G6, FA2H, SCP2, CRAT, and C19orf12, that participate in lipid metabolism; PANK2 and COASY are key enzymes in CoA biosynthesis; WDR45 and ATP13A2 that participate in autophagy process; FBXO7 involved in ubiquitin–proteasome protein-degradation pathway and mitophagy; REPS1 and AP4M1 are associated with vesicle trafficking; finally, DCAF17 and GTPBP2 with unclear function. Reference Hayflick, Kurian and Hogarth4,Reference Levi and Tiranti83,Reference Correa-Vela, Lupo and Montpeyó87
Treatment
Treatment of NBIA has been mostly symptomatic, supportive, and palliative for many decades. Drugs like tetrabenazine, baclofen, anticholinergics, antidopaminergic agents, and levodopa are partially useful in the treatment of major symptoms of NBIA. Reference Schneider3,Reference Cheon, Kim and Igarashi89 However, there has been a great struggle to implement disease-modifying drugs for NBIA patients. Chelation therapy for iron has been the major subject of research in this regard, but disease-specific treatment measures are being deployed such as (1) delivering alternative substrates downstream of the defective enzyme, (2) activating isoenzymes, (3) counteracting downstream cellular effects, (4) gene therapy, and (5) enzyme replacement therapy. Deferiprone, an oral iron chelator, which crosses the blood-brain easily is the most common chelating agent investigated in NBIAs. Despite the numerous investigations conducted for PKAN the results regarding deferiprone efficacy in this NBIA is still controversial. There are few reports of chelation therapy in PLAN, BPAN, MPAN and neuroferritinopathy with mixed results and data in these subtypes are scarce. Regarding aceruloplasminemia, due to iron deposition in other organs alongside the brain, the number of studied cases and the results are much more promising. Beyond the iron chelation many studies in preclinical or clinical phases are investigating the effect of specific genetic or enzymatic interventions on different NBIA subtypes as alluded above. Reference Iankova, Karin, Klopstock and Schneider90
NBIA in IRAN
There are more than 20 studies about NBIAs in Iran (Table 1); however, there is a lack of exact information about the prevalence of NBIAs among this population. Until the time of this paper, 39 families with 53 affected individuals are reported in Iran. Respectively, there were 23, 5, 3, 1, 3, 3, and one family with PKAN, MPAN, PLAN, JABELS, BPAN, Kufor-Rakeb and WSSs in Iran (Figure 4). Reference Jaberi, Rohani and Shahidi74,Reference Haeri, Haji Akhoundi, Alavi, Abdi and Rohani91–Reference Eslami, Razmeh, Abdollahi, Ghurchian and Maher109 Like the other countries, the most common form of NBIA was PKAN accounting for more than half of the cases (59%) (Figure 4). A high percentage of the PKAN may be due to the easy diagnostics procedures; the specific pattern on MRI, which is the “eye of tiger” sign. MPAN was the second most common form (13%) of NBIA among the Iranian population. While based on investigations in other countries, the PLAN seems to be more prevalent and accounts for about 20% of NBIA cases, it was accounted for only 8% of the Iranian NBIA patients. The low frequency of PLAN may be explained by several reasons: (1) lower prevalence in Iran; (2) less diagnosis (milder phenotypes in atypical forms and overlaps with other neurodegenerative disorders); and (3) failure to report cases. In the authors’ opinion, one main reason is the lack of referring these cases from neuropediatric clinics to movement disorders centers including ours.
Table 1: Iranian reported NBIA affected individuals

NA: not available; M: male; F: female; Hom: homozygous; Het: heterozygous; KRS: Kufor-Rakeb syndrome; Ref: reference; WSS: Woodhouse–Sakati syndrome.

Figure 4: Prevalence of NBIA subtypes among the Iranian population.
BPAN and KRS had prevalences equal to PLAN cases. The reports of KRS from Iran (eight patients from three families) look very interesting since there are few case reports from all over the world. Interestingly, JABELS was reported from Iran for the first time, and subsequently, mutations in gene GTPBP2 were reported in three unrelated Arab families from other countries, with a wide range of neurologic and non-neurologic features. Reference Bertoli-Avella, Garcia-Aznar and Brandau110 The first only case of WSS in Iran has also been reported recently. Reference Haeri, Haji Akhoundi, Alavi, Abdi and Rohani91
Also, we found one case of FAHN and a family affected by aceruloplasminemia with two affected siblings who have not been reported yet. There are no reports of neuroferritinopathy and CoPAN in the Iranian population so far, meaning that these forms are very rare (Table 1).
Conclusion
Due to phenotype and genotype heterogeneity in NBIAs, diagnosis is challenging and demanding. In many cases, due to lack of specific iron accumulation patterns on brain MRI, misdiagnosis is inevitable, while others have overlapping clinical symptoms. The aforementioned explanations plus unreported cases might result in lower prevalence compared to the real prevalence in some populations. In some cases, there are mutations in already-known genes related to NBIA without evidence of iron accumulation; on the contrary, some cases show sensible iron accumulation in the brain but no variation in already-known genes, making this disease far more complicated. Despite the undiscovered points about NBIA, the issue that there is no relationship between many NBIA genes and iron deposition increases the complexity of NBIAs.
This shows regarding the identification of molecular and pathophysiological aspects of NBIA, we are still at the beginning of the road to understand the real nature of this entity, and more investigations are needed. Since the pattern of inheritance in NBIA is mainlyAR, in populations with consanguine marriage like Iran, it is likely to find novel NBIA genes and the consanguine marriages may explain the higher prevalence of NBIA in Iran in comparison with the worldwide prevalence. Recent findings of NBIA suggest that other HSPs and Parkinson’s disease genes might be also involved in NBIAs, so it indicates that some NBIA genes might not be reported yet.
Acknowledgements
We acknowledge the Genetics Research Center at the University of Social Welfare and Rehabilitation Sciences for funding the research.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Statement of Authorship
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; and (3) Manuscript: A. Writing of the first draft, B. Review and Critique. Reza Hajati: 3B. Fardad Danaee Fard: 3B. Maziar Emamikhah: 2C, 3B. Mohammad Rohani: 1A, 1B and 3B. Afagh Alavi: 1A, 1B and 3B.





