


Most mammals synthesise ascorbic acid in the liver to fulfil their nutritional requirements. The last steps of the biosynthesis of ascorbic acid involve the enzyme l-gulonolactone oxidase(Reference Nishikimi and Yagi1). In species such as humans and guinea pigs, the gene for l-gulonolactone oxidase is mutated, and therefore the bioproduction of ascorbic acid is absent(Reference Burns2). Humans are completely dependent on acquiring exogenous ascorbic acid from the diet.
Ascorbic acid is essential for the synthesis of collagen in humans, and deficiency of ascorbic acid can lead to the disease scurvy where collagen molecules are destabilised, giving rise to various symptoms including haemorrhage, myalgia and anaemia. A retrospective study by Olmedo et al. (Reference Olmedo, Yiannias and Windgassen3) has shown that in modern industrialised societies, scurvy is mostly a comorbid condition in already ill patients or occurs as a complication of disorders affecting nutrition in general. The chemical structure of ascorbate makes it a good electron donor/acceptor, and it is this antioxidative/pro-oxidative property for which ascorbic acid is best known for. In the plasma, ascorbate is the primary antioxidant and is required to prevent the peroxide-mediated oxidation of LDL(Reference Jialal and Grundy4). Ascorbic acid is also one of the best enhancers of the intestinal absorption of Fe in humans(Reference Hallberg, Brune and Rossander5). The Fe uptake-enhancing effect of ascorbic acid has been shown in cell studies to be due to extracellular effects(Reference Bridges and Hoffman6). In a previous study in our laboratory, it was shown that ascorbic acid also affects the regulation of Fe uptake by intracellular mechanisms(Reference Scheers and Sandberg7).
In the present study, we examined the regulation of the Na-dependent vitamin C transporter 1 (SVCT1) in human intestinal Caco-2 cells in the presence or in the absence of Fe, in order to elucidate whether Fe status affects the uptake of ascorbate. Preliminary data suggest a synergistic relationship for the uptake and utilisation of these two nutrients. A possible intermediary in the crosstalk between Fe and ascorbate is protein kinase C (PKC), which has been found to play a role in the metabolism of both Fe and ascorbic acid.
PKC is an intracellular kinase that is a mediator in several intracellular signal transduction cascades. It is activated by second messengers such as cyclic AMP and diacylglycerols. Various isoforms of PKC exist, and the β-isoform has been shown to be activated by ferric iron bound to transferrin in several cell types(Reference Alcantara, Obeid and Hannun8). Activated PKC-β phosphorylates the Fe-responsive element-binding protein(Reference Eisenstein, Tuazon and Schalinske9), which suggests that it is involved in the regulation of the affinity of Fe-responsive element-binding protein for its substrates transferrin, Nramp2 and ferritin mRNA.
Ascorbate is actively transported into the cell interior by two transporters: SVCT1 and SVCT2. SVCT1 is believed to be the predominant isoform in the human intestine. This dominance is supported by indications that SVCT1 mRNA expression is down-regulated by the presence of ascorbate in the intestinal lumen, while SVCT2 is unaffected(Reference MacDonald, Thumser and Sharp10). A second route exists for indirectly increasing intracellular ascorbate levels. Facilitated diffusion of oxidised ascorbate, dehydroascorbate, occurs through the GLUT1, -3 and -4 transporters(Reference Rumsey, Kwon and Xu11, Reference Rumsey, Daruwala and Al-Hasani12). Dehydroascorbate is then reduced to ascorbate inside the cell or transported across the basal membrane into the circulation by means of these glucose transporters. It has been shown that the ascorbate transporter SVCT1 is exclusively expressed on the apical membrane of Caco-2 cells, and therefore does not mediate the basal transport of ascorbate(Reference Maulén, Henriquez and Kempe13).
The presence of SVCT1 transporters in the apical membrane has been shown to be regulated by PKC in transfected monkey kidney (COS-1) cells(Reference Liang, Johnson and Ma14). In this cell type, activated PKC (by phorbol 12-myristate 13-acetate) leads to a decrease in ascorbate uptake that is suggested to be caused by a down-regulation of SVCT1 transporters at the cell surface. To our knowledge, there are no studies on the effects of ascorbic acid on PKC activity. Our hypothesis is that ascorbic acid activates PKC, which leads to the down-regulation of SVCT1 and subsequently lowers the uptake of ascorbate. Ascorbate has been previously shown to down-regulate SVCT1 expression in Caco-2 cells(Reference MacDonald, Thumser and Sharp10), which was also the case in the present study. The major hypothesis of the present study is that Fe affects the uptake of ascorbate. We have focused on evaluating the involvement of the SVCT1 protein, and we have attempted to investigate whether PKC could be responsible for the observed effects.
Materials and methods
Materials
Plastics for cell cultivation were produced by Corning (San Fransisco, MA, USA). Media, supplements and other reagents for culture maintenance were produced by PAA Laboratories (Pasching, Austria). Reagents were obtained from Sigma-Aldrich (Schnelldorf, Germany), if otherwise not stated. For the experiments, ascorbic acid was used in the form of sodium ascorbate, and Fe was added as Fe(II)Cl2.4H2O.
Primary and secondary antibodies
Antibodies used in the present study were commercially produced, tested and recommended for the application. Rabbit anti-human SVCT1 was produced by Alpha Diagnostic (San Antonio, CA, USA). Specificity was successfully tested by blocking the SVCT1 antibody recognition site with a fifteen-amino acid immunising peptide identical to a portion of the binding region in the cytoplasmic domain of human SVCT1. According to the manufacturer, there is no significant homology with related transport proteins for the control peptide. For the calculation of standard curves in the ELISA experiments, ferritin was detected in samples of known concentration by a rabbit anti-human ferritin antiserum (Sigma-Aldrich). In the ELISA, the primary rabbit IgG were detected by a goat anti-rabbit IgG β-lactamase tumour endothelial marker 1 (TEM)-1 conjugate (Invitrogen, Eugene, OR, USA). For Western blots, the secondary antibody was a goat anti-rabbit IgG conjugated with horseradish peroxidase (Bio-Rad, Sundbyberg, Sweden).
Cell line and cell culture
Caco-2 cells (HTB-37) were purchased from the American Type Culture Collection (Rockville, MD, USA) at passage 19. Stock cultures were maintained in Dulbecco's modified Eagle's medium supplemented with heat-inactivated fetal bovine serum (FBS; 16 % (v/v)) and non-essential amino acids (0·9 % (v/v)). The cell cultures were grown at 37°C in 95 % humidified air and 5 % CO2. At passages 32–41, the cells were seeded in twelve-well plates with Transwell® polycarbonate inserts at 60 000 cells/insert. All experiments were initiated 14 d after seeding. No antibiotics or antifungal agents were used.
Cell experiments for the measurement of ascorbic acid uptake
At 12 d after seeding, the medium was exchanged for minimal essential medium (MEM) with Earle's salt (E15-825; PAA) supplemented with 2 % (v/v) FBS. At 24 h before the experiments, the cells were treated in the basal compartment with MEM-FBS, unsupplemented or supplemented with Fe (10 μmol/l). Fe is readily absorbed from this compartment into the cell. The experiments were initiated by washing the cells in PBS followed by incubation with Hank's balanced salt solution supplemented with ascorbic acid (150 μmol/l) in the apical compartment and Hank's balanced salt solution with Fe (10 μmol/l) in the basal compartment. Care was taken to make sure that there was no contact between the Fe- and ascorbic acid-containing media. The cells were incubated with these media for 2 h, after which the apical and basal media were collected for ascorbic acid analysis. The cells were lysed in radio-immunoprecipitation assay buffer and divided into aliquots for total protein and ascorbic acid analyses. All samples for ascorbic acid analyses were diluted to a final concentration of meta-phosphoric acid (3 % (v/v)), Tris (carboxyethyl) phosphine hydrochloride (0.312 mmol/l) and EDTA (1 mmol/l). The samples were also diluted without Tris (carboxyethyl) phosphine hydrochloride to distinguish between ascorbic acid and dehydroascorbic acid(Reference Lykkesfeldt15). The ascorbic acid analyses were run by HPLC. A Jasco system (Jasco Corporation, Tokyo, Japan) with a Jasco As-2080 Plus pump and autosampler Jasco As-2057 was employed. The samples were separated using an Aquasil C18, 3 μm and 150 × 4·6 mm column (Thermo Electron Corporation, Marietta, OH, USA). The flow rate was 0·4 ml/min. The mobile phase was NaH2PO4 (50 mmol/l) adjusted to pH 2·8 with H3PO4 (1·5 mol/l), EDTA (200 mg/l) and methanol (2 % (v/v)). Electrochemical detection was carried out using a Decade II system; Antec Leyden in DC mode (flow cell: VT-03, glassy carbon WE, 3 mm; E-cell: 800 mV v. Ag/AgCl/KCl; I-cell: 50 nA). Signal analysis was conducted using Jasco ChromPass software (Jasco Corporation).
Cell experiments for the investigation of protein kinase C involvement and inhibition of sodium-dependent vitamin C transporter 1
The basic procedure was as described in the previous section with a few exceptions. The Fe concentration was 10 or 20 μmol/l. Cells were treated with the PKC inhibitor Go 6983 (5 μmol/l), the PKC activator phorbol 12-myristate 13-acetate at 1 μmol/l or the SVCT1 transport inhibitor quercetin dihydrate (50 μmol/l) for 1·5 h before the ascorbate addition. The co-incubation lasted for 1·5 h. The inhibitors and the activator were all dissolved in dimethyl sulphoxide. The total concentration of dimethyl sulphoxide in the wells was 0·01 %. Dimethyl sulphoxide (0·01 %) was also given to the cells as a control. After the co-incubations, the cells were lysed, and intracellular ascorbate/dehydroascorbate levels were analysed by HPLC. The apical media were analysed to differentiate between ascorbate and dehydroascorbate levels at the start (t = 0 h) and at the end (t = 1·5 h) of the incubation. This was done to predict whether, to what extent, the uptake could be mediated by SVCT1 (ascorbate) or by GLUT (dehydroascorbate).
Cell experiments for the measurement of sodium-dependent vitamin C transporter 1 expression
At 12 d post-seeding, the medium was exchanged for MEM supplemented with 2 % (v/v) FBS (MEM-FBS). For these experiments, the MEM used had been verified to contain no ascorbic acid or Fe by analyses in our laboratory. The study design was as described in a previous publication(Reference Scheers and Sandberg7) with some modifications. Briefly, on day 13, the cells were treated on the apical side with either unsupplemented MEM-FBS (Expts 1, 2 and 5) or MEM-FBS supplemented with ascorbic acid (150 μmol/l; Expts 3, 4 and 6). Half the plates were also treated on the basal side of the membrane with MEM-FBS supplemented with ascorbic acid (150 μmol/l; Expts 2–6). The experimental set-up is described in Table 1.
Table 1 Description of Expts 1–6*

* The experiments started at t = 0 h and were terminated at t = 21 h. For half the cells, lysis occurred immediately at t = 21. The other half was given minimal essential medium-fetal bovine serum and put back into the incubator for another 22 h and lysed at t = 43 h.
Before incubation with Fe in Expts 4–6, the apical medium was exchanged for MEM, supplemented as shown in Table 1. The integrity of the monolayer was verified by transepithelial electric resistance. In Expt 6, the ascorbic acid-treated cells were carefully washed in PBS to remove all apical extracellular ascorbic acid before Fe incubation experiments. This ensured that only intracellular ascorbate was present. In Expts 4–6, the cells were incubated with Fe for 2 h on a rotary shaker (25 rpm and 37°C) after which they were washed in PBS. Cells were harvested immediately from half the plates. Fresh medium (MEM-FBS) was added to the rest of the plates, which were incubated for a further 22 h before harvest.
Harvesting of Caco-2 cells for protein analysis
Before harvest, the medium was aspirated and the cells were washed in PBS. Cells were lysed either immediately (t = 21) or 22 h (t = 43) after the removal of test solutions. Cells were lysed in cold radio-immunoprecipitation assay buffer (Sigma-Aldrich) containing an EDTA-free protease inhibitor cocktail (40 μl/ml; Roche, Basel, Switzerland). All lysed cell samples were handled in a cold environment. The lysates were divided into aliquots and frozen at − 80°C for later analysis. Total protein content was estimated in aliquots of the individual lysates by the bicinchoninic acid assay (Pierce, Stockholm, Sweden). A commercial ferritin ELISA kit (Calbiotech, Spring Valley, CA, USA) was used for the detection of intracellular ferritin levels.
Direct capture ELISA for the quantification of sodium-dependent vitamin C transporter 1 expression
The SensiFlex™ ELISA development kit (Invitrogen) was used according to the manufacturer's instructions. The concentrations of antigen and primary antibodies (0·5 μg/well) were optimised to maintain a relatively low total antigen concentration in order to minimise competition for binding sites. A microplate was coated with samples diluted (1:20) in NaHCO3 (50 mmol/l) and was then incubated overnight at 4°C on a rotary shaker (20 rpm). The next day, the plate was drained, and the wells were washed three times with PBS-Tween (200 μl). After draining, PBS-bovine serum albumin (200 μl) was added to the wells to block unspecific binding of the antibody to the plate. The plate was again incubated overnight at 4°C while being gently agitated. On the third day, the plate was drained and washed three times in PBS-Tween (200 μl). After thorough draining, the wells were treated with the primary antibody (0·5 μg) diluted in NaHCO3 (100 μl of 50 mmol/l) and then incubated for 30 min at room temperature. The plate was drained and washed three times in PBS-Tween (200 μl). The secondary antibody (goat anti-rabbit IgG conjugated with β-lactamase) was diluted in PBS-bovine serum albumin (1 μg in 100 μl) and added to each well. The plate was incubated for 30 min at room temperature, protected from light. After incubation, the plate was drained and washed three times in PBS-Tween (200 μl).
Fluorocillin Green reagent (100 μl of 9 μmol/l) was added to each well and incubated for 30 min, protected from light. β-Lactamase activity was then detected using a fluorescence microplate reader Sapphire 2 (Tecan GmbH, Salzburg, Austria). The excitation and emission wavelengths were 490 and 525 nm, respectively. The elapsed time between the end of the incubation step and plate reading was kept constant for all experiments. The concentration of SVCT1 was then calculated from the standard curve and normalised against the total protein content of each sample. All values were within the range of the standard curve. SVCT1 expression was also analysed by Western blot using the same method as reported previously(Reference Scheers and Sandberg7) in order to verify the results of the ELISA.
Statistics
Experiments were performed on three batches of cells within each run, and every run was repeated on three different occasions. In addition, each trial of SVCT1 and ferritin expression studies was duplicated in order to lyse a set of experiments at two time points (t = 21 and 43 h). With this design, each triplicate of control cells (ascorbate or no ascorbate) could always be compared against another triplicate from the same run, giving a total of eighteen samples of control cells, verifying the reproducibility of the method. All values are reported as means of nine individual samples with their standard errors (n 3). The significance of differences between treatments and their control was analysed by Student's unpaired two-tailed t test using Microsoft Office Excel. Differences were considered significant at P < 0·05.
Results
Ascorbic acid uptake
Ascorbic acid uptake across the apical membrane was increased in cells supplemented with Fe (FeCl2·4H2O) in the basal extracellular compartment compared with unsupplemented cells (difference of 285 %, P = 0·03; Fig. 1). Transport of ascorbic acid across the basal membrane was also measured. The percentage transport, calculated from the intracellular ascorbate levels, across the basal membrane decreased in cells incubated with basal Fe compared with those not treated with Fe. However, the absolute amount of ascorbic acid transported was unchanged (0·11 v. 0·10 μg ascorbate/mg protein, P>0·05; Fig. 1). The latter result could indicate that ascorbic acid transport was already at maximum velocity, and no further increase was possible. However, these experiments need to be repeated with non-saturable concentrations of ascorbic acid, before any firm conclusions can be drawn.

Fig. 1 Cellular uptake and transport of ascorbic acid. The cells were incubated with Fe in the basal chamber for 24 h followed by apical treatment with ascorbic acid (150 μmol/l) lasting for 2 h. Values are means, with standard errors represented by vertical bars (n 3). Ascorbate uptake (![]() ) was defined as the measured concentration of ascorbate/dehydroascorbate in the cell lysates. Ascorbate transport (
) was defined as the measured concentration of ascorbate/dehydroascorbate in the cell lysates. Ascorbate transport (![]() ) was estimated by measuring the concentration of ascorbate/dehydroascorbate in the basal medium.
) was estimated by measuring the concentration of ascorbate/dehydroascorbate in the basal medium.
During the ascorbate uptake experiments, the apical medium was analysed for ascorbate and dehydroascorbate. The first set of samples (10 μl) was withdrawn immediately after the addition to the cells, and a second set of samples (10 μl) was withdrawn after 1·5 h of incubation. The results showed that the initial samples contained 20 (se 3) % dehydroascorbate of the total ascorbate/dehydroascorbate concentration. After 1·5 h in the incubator, the dehydroascorbate content was increased to 26 (se 4) % of the total concentration of the two measured species. The difference between the two time points was not statistically significant. It is worth noting that the total concentration of ascorbate and dehydroascorbate was reduced by 18 (se 5) % during the 1·5 h of incubation.
Sodium-dependent vitamin C transporter 1 expression
When ascorbate-deficient cells were exposed to apical Fe (Expt 5), SVCT1 protein expression increased by 23·7 (se 5) % (P = 0·02) v. control cells (Expt 1) at t = 43 h. This increase was detectable at t = 21 h, 2 h after the addition of Fe (9 (se 2) %, P = 0·05; Fig. 2, Expt 5). This result suggests that the regulation of SVCT1 expression by Fe could be a relatively fast response compared with, for example, ferritin expression. Ferritin is regulated at the mRNA level by the Fe-responsive element/Fe-regulatory protein pathway, where the effect of Fe administration is not apparent after 2 h. SVCT1 mRNA does not show any significant sequence similarities with iron-responsive element (IRE) sequences from ferritin or ferroportin mRNA according to the Internet-based nucleotide Basic Local Alignment Search Tool (Blastn; National Center of Biotechnology Information, Bethesda, MD, USA). SVCT1 expression was significantly decreased 2 h after the apical addition of ascorbic acid to ascorbic acid-deficient cells (22·4 (se 2) %, P = 0·05; Fig. 2, Expt 3). However, after 22 h with unsupplemented medium, the levels of SVCT1 were normalised, which further supports a fast regulatory mechanism.
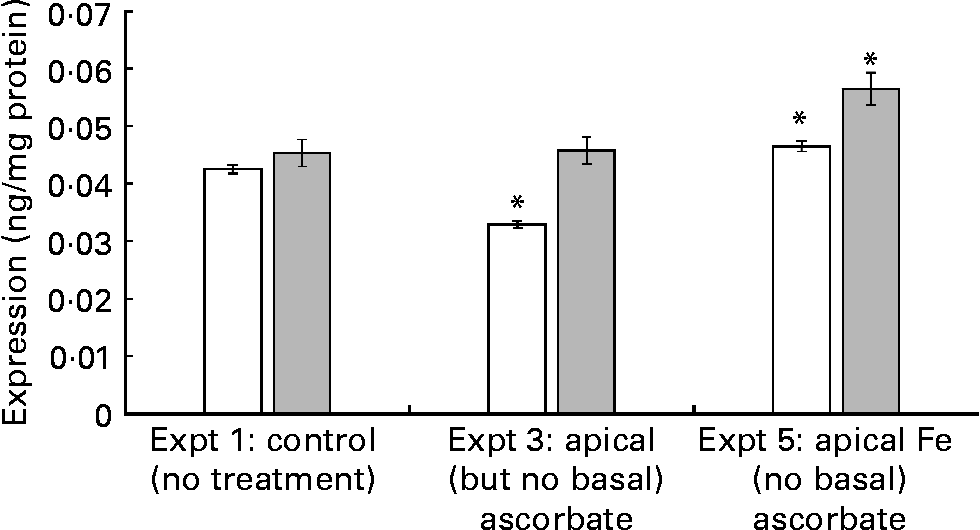
Fig. 2 Na-dependent vitamin C transporter 1 expression in ascorbate-deficient cells. The cells were incubated with either ascorbate (150 μmol/l, 21 h; Expt 3) or Fe(II)Cl2.4H2O (20 μmol/l, 2 h; Expt 5). After finishing the incubation, the cells were lysed immediately (t = 21 h, □) or 22 h later (t = 43 h, ![]() ). The experimental design was according to Table 1, except that no basal ascorbate was added. Values are means of nine samples, with standard errors represented by vertical bars (n 3). * Mean values were significantly different from that of the control cells (P < 0·05).
). The experimental design was according to Table 1, except that no basal ascorbate was added. Values are means of nine samples, with standard errors represented by vertical bars (n 3). * Mean values were significantly different from that of the control cells (P < 0·05).
The ascorbic acid transporter protein SVCT1 was slightly up-regulated (10 (se 2) %, P = 0·05) in the presence of basal ascorbic acid as observed at t = 21 h (Fig. 3, Expt 2). Caco-2 cells with replete ascorbic acid levels did only moderately respond to apical treatments with ascorbate and/or Fe ( − 5·25 (se 4) %, Fig. 3, Expt 4, t = 43 h, P = 0·05; Expts 5–6; no significant changes; P>0·05). The relatively weak response (Expts 4–6) suggests that the effect of Fe on ascorbic acid uptake is dependent on the availability and/or cellular status of ascorbic acid. However, in Expt 4 where the change was significant, the ferritin levels were higher than in Expts 5–6. It should also be remembered that a relatively small change in total SVCT1 levels might give rise to a considerably higher response in ascorbic acid uptake.

Fig. 3 Ferritin and Na-dependent vitamin C transporter (SVCT1) expression in ascorbate-replete cells. Ferritin and SVCT1 protein expression in Caco-2 cells grown on inserts. Ferritin values have been divided by 500 (□). The medium in the basal chambers was supplemented with ascorbate (150 μmol/l, Expts 2–6). See Table 1 for an explanation of the different combinations of Fe and ascorbate used in each experiment. After finishing 2 h of Fe incubation, the cells were lysed immediately (SVCT1 at t = 21 h, ![]() ) or 22 h later (SVCT1 at t = 43 h,
) or 22 h later (SVCT1 at t = 43 h, ![]() ). Values are means of nine samples, with standard errors represented by vertical bars. * Mean values were significantly different from that of the control cells (with basal ascorbate; P < 0·05).
). Values are means of nine samples, with standard errors represented by vertical bars. * Mean values were significantly different from that of the control cells (with basal ascorbate; P < 0·05).
To verify the ELISA results, SVCT1 expression was also estimated by Western blot analyses for random samples of treatment and controls. The Western blots showed an increase in SVCT1 expression when ascorbic acid-depleted cells exposed to apical Fe were analysed at t = 21 h and had an even greater increase at t = 43 h (data not shown), verifying the trends measured by ELISA (Fig. 2, Expts 1 and 5). The correlation between the increased ascorbate uptake and the increased SVCT1 expression was evidenced by treating the cells with the reversible SVCT1 inhibitor quercetin. Quercetin (50 μmol/l) decreased the uptake of ascorbate in Fe-treated cells by 68 % (1·34 (se 0·07) v. 0·4 (se 0·05) μg/mg protein, P = 0·00001).
Possible involvement of protein kinase C
PKC inhibition by Go 6983 did not increase ascorbate uptake. Indeed, Go 6983 decreased ascorbate uptake by 20 (se 5) % (P = 0·04) regardless of Fe status. PKC activation by phorbol 12-myristate 13-acetate decreased ascorbate uptake significantly, as expected, by 84 % (data not shown) irrespective of Fe status, suggesting that the Fe- stimulatory effect of ascorbate uptake is not facilitated by changes in PKC-mediated trafficking. Phorbol 12-myristate 13-acetate has been shown to affect PKC-stimulated trafficking from the cell surface to the cytosol(Reference Liang, Johnson and Ma14) and vice versa(Reference Lin and Chen16). The results of the present experiments do not necessarily exclude the involvement of PKC isoforms in Fe-stimulated ascorbate uptake. Further studies need to be conducted.
Intracellular ferritin expression
Caco-2 cells grown on inserts with ascorbate in the basal compartment showed significantly higher baseline ferritin expression (22 (se 7) %; P = 0·05) compared with cells grown without ascorbate in the basal chamber (Fig. 3, Expts 1 and 2). In contrast, when cells basally replete for ascorbate were treated with ascorbate at the apical cell membrane, ferritin expression was significantly down-regulated by 37 (se 1) % (P = 0·04; Fig. 3, Expt 3). The results for Expts 1–3 (Fig. 3) are in accordance with the previous data from our laboratory (N. M. S. and A.-S. S., unpublished results), suggesting that the effect of ascorbate at the apical membrane is different from the effect at the basolateral surface. The apical membrane was found to be impermeable to ascorbate leaving the cell. The findings were confirmed by the analysis of the basal, intracellular and apical compartments of ascorbate-replete control cells. HPLC analysis showed that only the apical medium was devoid of ascorbate.
In the present study, when the Caco-2 cells were supplied with basal ascorbate, there was no observable difference in ferritin levels between cells that were treated with apical Fe (Fig. 3, Expt 5) and those treated with intracellular ascorbate and apical Fe (Fig. 3, Expt 6). Due to the earlier observed transport of ascorbate from the basal chamber to the cell interior, this outcome was expected as the treatments were effectively the same. The basal influx of ascorbate is likely to be mediated by the GLUT 1, -3 and -4 transporters, which normally function as glucose transporters but which also transport dehydroascorbate(Reference Maulén, Henriquez and Kempe13).
Discussion
There have been many studies on Fe metabolism and the importance of maintaining an adequate Fe status in humans. For many years, there have been general recommendations to ingest ascorbic acid in order to stimulate Fe uptake in the small intestine. In the present study, we have demonstrated the involvement of Fe in the up-regulation of the vitamin C transporter protein SVCT1 in ascorbate-depleted human intestinal Caco-2 cells. We have also shown that ascorbic acid uptake was increased if intracellular Fe levels were increased, and we have proved the correlation between the increased expression of the SVCT1 transporter and ascorbic acid uptake. These results are important as they change our view of the relationship between ascorbic acid and Fe intake.
A study by Núñez et al. (Reference Núñez, Tapia and Rojas17) has shown that the extracellular availability of Fe determines whether the Fe influx and efflux transporters Nramp2 and ferroportin will localise to the apical or the basal membrane in Caco-2 cells. The consequence of this mechanism for the present study is that both apical and basal supplementation of Fe will contribute to an increase in intracellular Fe concentration caused by apical or basal influx via Nramp2. The basal influx of Fe is also affected by transferrin originating from the FBS-supplemented medium.
In the present study, total cellular SVCT1 expression increased by approximately 24 %, when the cells were exposed to Fe. This measurement was made 22 h after an Fe uptake experiment that lasted for 2 h in total. In contrast, an increase in ascorbic acid uptake of 285 % was observed after basal Fe supplementation for 24 h. This difference in experimental design could possibly explain the very pronounced effect that Fe supplementation had on ascorbic acid uptake compared with the effect on SVCT1 protein expression. There could also be other regulatory mechanisms that affect the effective uptake of ascorbic acid, such as increased transport capacity or intracellular redistribution, which is a common mechanism for other transmembrane transport systems, for instance Nramp2 and ferroportin-1 as described earlier(Reference Núñez, Tapia and Rojas17). However, we observed an effect on the total protein expression of SVCT1 when cells were exposed to Fe.
A study by MacDonald et al. (Reference MacDonald, Thumser and Sharp10) has shown that an increase in the extracellular ascorbate level leads to the down-regulation of SVCT1 mRNA in Caco-2 cells. In the present study, we found an initial decrease, on the protein level, that normalised after approximately 24 h. The results reported by MacDonald et al. might lead to the conclusion that the regulatory effect of ascorbate on SVCT1 takes place at the mRNA level. As observed in the present study, the cellular response at the protein level occurred so rapidly that it seems likely that the regulation of SVCT1 also occurs at the translational level.
We conclude that Fe stimulates the uptake of ascorbate and the expression of the SVCT1 transporter protein in human Caco-2 cells. This stimulatory effect is greater when the ascorbic acid level in the cell is low.
Acknowledgements
The present study was funded by the Swedish Council for Environment, Agricultural Sciences and Spatial Planning (Formas; grant no. 222-2004-1889) and the Dr Per Håkansson Foundation. The authors state that there are no conflicts of interest. We are grateful to Nils-Gunnar Carlsson for expert technical assistance regarding the ascorbic acid analyses. N. M. S. provided the hypothesis, designed the experiments and performed the study. A.-S. S. contributed to the present study through planning and supervision of the entire work.




