



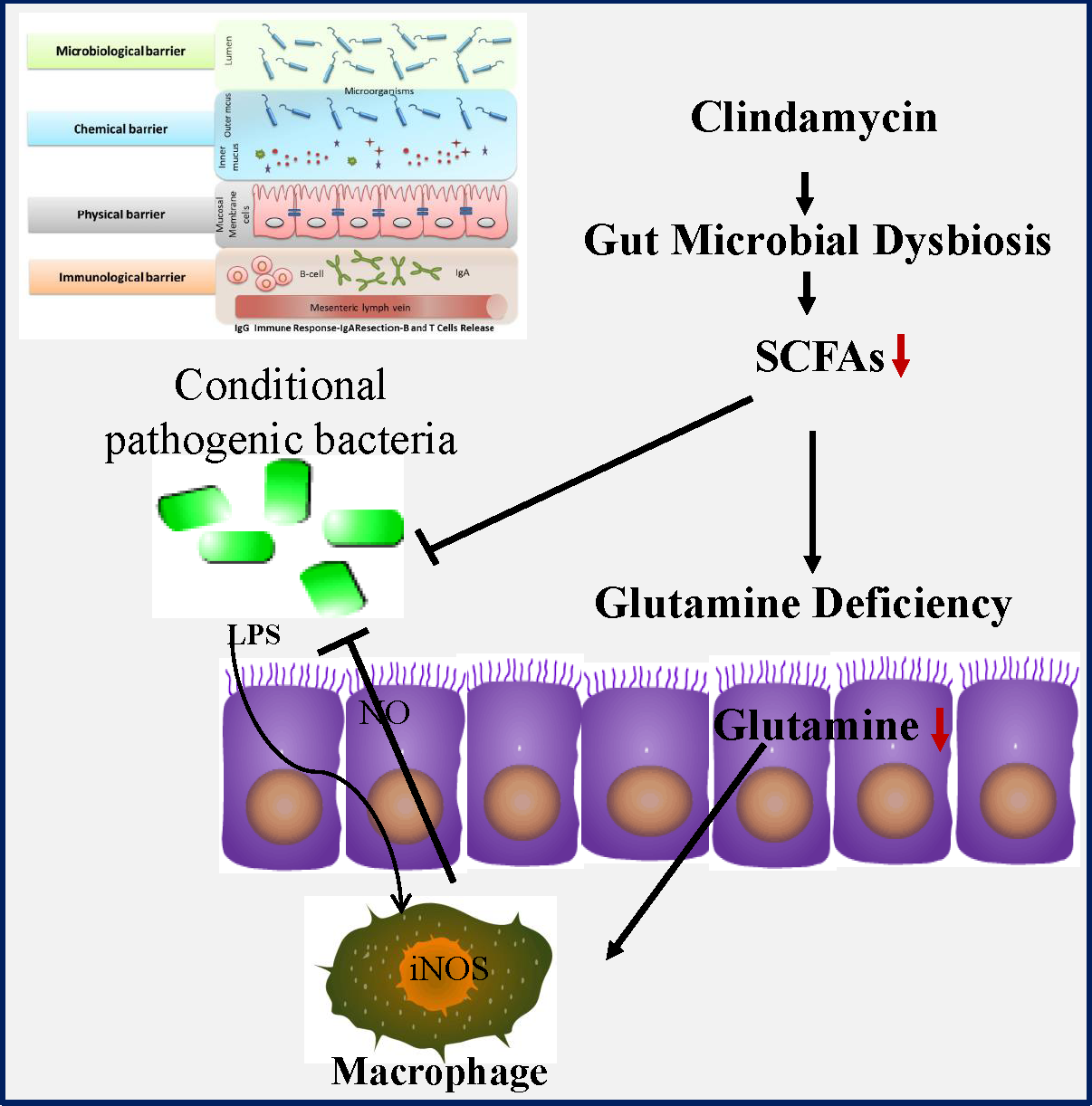
Humankind has been endowed with antibiotics as the most powerful weapon against bacterial infection, but their use is often limited by antibiotic-associated diarrhoea (AAD)(Reference Barbut and Meynard1–Reference Hempel, Newberry and Maher3). Occurring in 30 % of patients, diarrhoea represents a major adverse effect of antibiotic use, but so far there is no effective therapeutic regimen available. AAD not only affects the patients’ quality of life but also incurs significant healthcare costs worldwide. AAD is normally a self-limiting disease upon the withdrawal of antibiotic, but it often leads to the incomplete course of antibiotic therapy as well as the following more frequent and severe nosocomial infection(Reference Hryckowian, Van Treuren and Smits4,Reference Zackular, Moore and Jordan5) . For example, it might facilitate infection by conditional pathogenic bacteria such as Clostridium difficile, Salmonella, Clostridium perfringens and Escherichia coli. Although almost all antibiotics might cause AAD, the risk is rather higher in broad-spectrum antibiotics such as aminopenicillins, cephalosporins and clindamycin. Considering their huge clinical benefit in infectious diseases control, those drugs will undoubtedly continue to be the most commonly prescribed medicine. Therefore, a medical regimen capable of preventing or reducing symptoms of AAD would be urgently needed.
Accumulating evidence indicates that gut microbiota might functionally mediate host energy homoeostasis(Reference Backhed, Ding and Wang6–Reference Lee and Hase8). Butyrate from bacterial origin represents a major fuel for colonocytes, while colonic epithelial cells of germfree mice might increase the compensatory use of glutamine(Reference Cherbuy, Darcy-Vrillon and Morel9,Reference Scheppach, Dusel and Kuhn10) . Glutamine which is the most abundant non-essential amino acid in human body may become a ‘conditionally essential’ amino acid, especially in the states of tissue repair or severe illness(Reference Scheppach, Dusel and Kuhn10,Reference Zhou, Costinean and Croce11) . Importantly, glutamine deficiency has been implicated in intestinal permeability by altering physical and immunological barriers(Reference Scheppach, Dusel and Kuhn10–Reference Zhou, Verne and Fields12). In previous studies, we noticed that antibiotic use greatly affected the abundance of the SCFA-producing strains as well as intestinal permeability(Reference Shi, Kellingray and Zhai13,Reference Shi, Zhai and Li14) . Collectively, all those findings suggested that glutamine deficiency might link antibiotic-induced dysbiosis and intestinal barrier dysfunction in AAD. Nonetheless, the potential protective effects of glutamine on AAD remain uncharacterised yet. In the present study, we investigated whether glutamine supplementation could ameliorate AAD progression as well as the underlying mechanism of actions.
Materials and methods
Materials, chemicals and reagents
Primary antibodies against cyclo-oxygenase-1 (COX-1, no. 4841), cyclo-oxygenase-2 (COX-2, no. 12282), inducible nitric oxide synthase (i-NOS, no. 13120), nuclear factor-kappa Bα (IkBα, no. 24358), Ser32-phospho-ikBα (no. 2859), PPAR-γ (no. 24358) and solute carrier family 1 member 5 (SLC1A5, no. 5345) were purchased from Cell Signaling Technology. Primary antibody against glutamine synthetase (GS, ab64613) was obtained from Abcam. Enzyme immunoassay kits for d-lactic acid (no. ml063046), lipopolysaccharides (LPS, no. ml037221), occludin (no. ml063481) and zonula occludens 1 (ZO-1, no. ml037693) determination were from Shanghai Enzyme-linked Biotechnology. All chemicals were from Sigma–Aldrich unless indicated otherwise.
Cell culture
Human colon cancer cell line (HCT116) and immortalised mouse mononuclear macrophages (RAW264.7) were obtained from the American Type Culture Collection and maintained following their instructions. Cells were cytogenetically tested and authenticated before frozen. Each vial of the frozen cells was thawed and maintained for about 2 months.
Cell growth assay
Cell growth was measured by MTS assay(Reference Li, Zhu and Chen15). HCT116 and RAW264.7 cells were seeded (1 × 103 cells per well in ninety-six-well plates) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4 mm of glutamine (glutamine-complete), 2 mm of glutamine (glutamine-deficient) or no glutamine (glutamine-free). After incubation, 20 μl of CellTiter96Aqueous One Solution (Promega Corporation) was added, and the optical density was finally determined at 492 nm.
Wound healing assay
Wound healing was measured by cell scratch assay(Reference Zykova, Zhu and Wang16). HCT116 cells (6 × 105) were seeded into a six-well plate, allowed growth to 80 % confluence and pretreated with mitomycin C (10 μg/ml) for 2 h before wound healing assay. A single scratch was made by pulling a plastic tip across the cells. After washing with PBS, cells were cultured in DMEM containing different glutamine. Photographs were taken at indicated times and the ‘healing area’ was measured by ImageJ.
Murine primary peritoneal macrophage study
Murine primary peritoneal macrophages were isolated as described(Reference Hoque, Farooq and Ghani17). Primary peritoneal macrophages were adjusted to 2 × 106 cells/ml in the tissue culture medium. After incubation for 2 h, the culture medium was changed to discard non-adherent cells. Isolated mouse primary peritoneal macrophages were used for subsequent macrophage polarisation by LPS (1 μg/ml) or IL-4 (20 ng/ml) stimulation for 8 h. Total RNA was extracted with TRIzol reagent, and real-time PCR was carried out. The primer sequences for PCR are summarised in online Supplementary Table S1.
RAW264.7 macrophage activation
The effect of glutamine on LPS-induced classical antimicrobial macrophage activation was performed as described(Reference Jantsch, Schatz and Friedrich18). RAW264.7 cells were seeded (5 × 105 cells) in 10-cm cell culture dish. When cells reached 80 % confluence, cells were cultured in RPMI 1640 medium containing glutamine at different doses. Two hours later, cells were stimulated or not with LPS (1 μg/ml). Supernatant fractions were collected for nitric oxide (NO) measurement, whereas cells were collected for Western blot. NO was measured as nitrite using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemicals). For E. coli infection, RAW264.7 cells were infected with E. coli CMCC44102 with a multiplicity of infection of 100 for 2 h(Reference Jantsch, Schatz and Friedrich18). After infection, cells were washed to remove extracellular bacteria. Gentamicin (100 μg/ml) was added to prevent replication of the remaining extracellular bacteria. Infection was finally terminated by cellular lysis (0·5 % Triton X-100 in PBS, 5 ml), and the number of intracellular bacteria was determined by serial dilution and subsequent plating on Müller-Hinton agar plates. Colony-forming units were enumerated after incubation overnight at 37°C. For determination of free E. coli in medium, bacteria and cells are co-cultured for total 6 h.
Bacterial growth assay
Bacterial growth was performed as described(Reference Wu, Esteve and Tremaroli19). E. coli CMCC44102 was seeded into 9 ml of sterile culture medium. After overnight culture, 200 ul of bacterial solution was transferred into a new sterile test tube and co-cultured with or without glutamine for another 12 h at 23°C. Their absorbance at 600 nm was detected per h.
Western blot analysis
Cells or colonic tissues were lysed in radio-immunoprecipitation assay (RIPA) buffer, and protein concentration of cell lysates was determined by Bradford assay. Protein samples (20 μg) were analysed by Western blot as described(Reference Li, Zhu and Chen15). Western blots were finally visualised using an enhanced chemiluminescence reagent (GE Healthcare). The bands were quantified by scanning densitometry, normalised with the β-actin and quantified by comparison with control(Reference Wang, Chen and Mao20).
Clindamycin-induced antibiotic-associated diarrhoea murine model
Six-week-old male C57BL/6 mice were purchased from Model Animal Research Center of Nanjing University and maintained under ‘specific pathogen-free’ conditions according to guidelines established by Institutional Animal Care and Use Committee, Jiangnan University (protocol number JN 20171015-1128). Mice were kept in an air-conditioned room with controlled temperature (22 ± 1°C), humidity (65–70 %) and day/night cycle (12 h light, 12 h dark). A preclinical mouse model of AAD was adopted as described(Reference Shi, Kellingray and Zhai13,Reference Shi, Zhai and Li14) , and the experimental protocol is shown in Fig. 1(a). In brief, mice were randomly assigned to five groups (n 10 per group) and fed ad libitum with a control diet (1022, Beijing HFK Bioscience Co. Ltd). To initiate AAD, mice were treated with clindamycin (250 mg/kg body weight) every day for 14 d. Glutamine (2 g/kg) or the vehicle (PBS) was administered using an intragastric tube at 0·1 ml per 10 g every day throughout the experiment. According to the standard scaling factors used by the Food and Drug Administration (FDA), the mouse-to-human scaling factor was 12. Considering the fact that the recommended dose of glutamine for patients weighing 70 kg is 12·5 g once a day, mouse equivalent dose was calculated as 2·1 g/kg. Body weight, water intake and food intake were recorded every 3 d. At the end of the experiment, faeces were collected from mice directly into microcentrifuge tubes and extracted DNA immediately or stored at a −80°C freezer. After euthanisation, mice organs and blood were collected. Blood was centrifuged at 2000 g for 15 min, and the supernatant fraction was designated as plasma. Plasma or colonic levels of glutamine, glutamic acid (Glu), arginine, asparagine and aspartic acid (Asp) were determined using HPLC as described previously(Reference Xue, Sawyer and Field21).
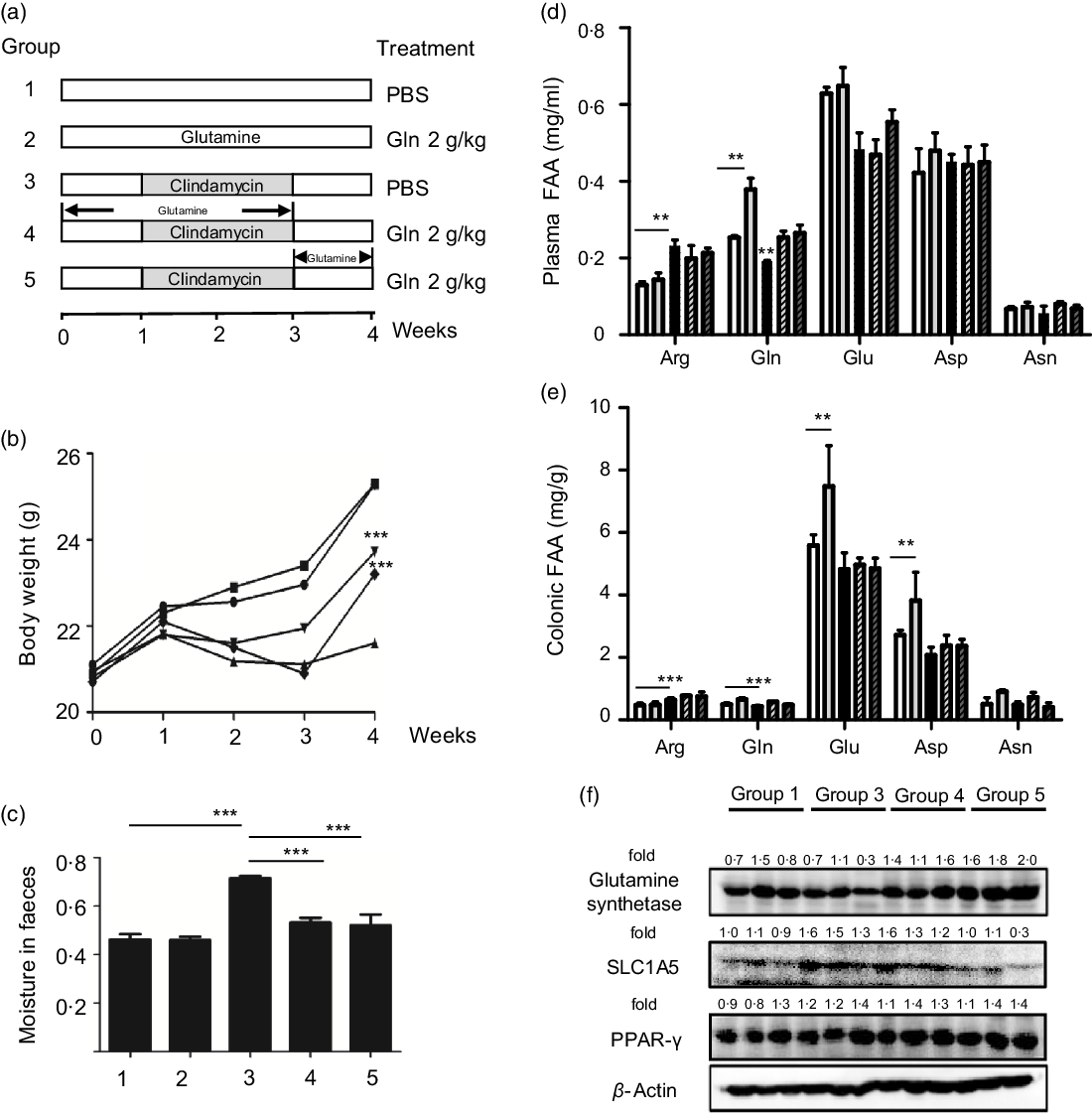
Fig. 1. Glutamine improved clindamycin-induced antibiotic-associated diarrhoea in mice. (a) Experimental design. (b) Body weight. ![]() , 1;
, 1; ![]() , 2;
, 2; ![]() , 3;
, 3; ![]() , 4;
, 4; ![]() , 5. (c) Moisture in faeces. (d) Plasma free amino acid (FAA) levels.
, 5. (c) Moisture in faeces. (d) Plasma free amino acid (FAA) levels. ![]() , 1;
, 1; ![]() , 2;
, 2; ![]() , 3;
, 3; ![]() , 4;
, 4; ![]() , 5. (e) Colonic FAA levels.
, 5. (e) Colonic FAA levels. ![]() , 1;
, 1; ![]() , 2;
, 2; ![]() , 3;
, 3; ![]() , 4;
, 4; ![]() , 5. Data are mean values and standard deviations (n 10 in each replicate). Significant difference compared with each respective vehicle group: ∗∗ P < 0·01, ∗∗∗ P < 0·001. (f) Effect of clindamycin on glutamine metabolism in colon mucosa tissues. The data represent results from three independent experiments. SLC1A5, solute carrier family 1 member 5.
, 5. Data are mean values and standard deviations (n 10 in each replicate). Significant difference compared with each respective vehicle group: ∗∗ P < 0·01, ∗∗∗ P < 0·001. (f) Effect of clindamycin on glutamine metabolism in colon mucosa tissues. The data represent results from three independent experiments. SLC1A5, solute carrier family 1 member 5.
Gut microbiota composition analysis
Total DNA of faeces samples was extracted using the Fast DNA Spin Kit for Soil (no. 6570200; MP Biomedical)(Reference Shi, Kellingray and Zhai13). The V4 region of the 16S rDNA was amplified by PCR and the primers were forward primer, 5′-AYT GGG YDT AAA GNG-3′, and reverse primer, 5′-TAC NVG GGT ATC TAA TCC-3′. The reaction mixtures were subjected to amplification with the following cycling conditions: initial denaturation at 94°C for 5 min followed by thirty cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 30 s. A final extension at 72°C for 5 min was performed. PCR products were gel-purified using Gene Clean Turbo. Products were prepared using TruSeqDNALT Sample Preparation Kit and sequenced on a MiSeq sequencer.
Faecal SCFA determination
Dried faeces were ground to powders and diluted with distilled water for ultrasonic treatment. After derivatisation, SCFA were measured by GC as described(Reference Shi, Zhai and Li14). Total SCFA concentration was calculated as the sum of acetic, propionic, butyric, isobutyric and valeric acid concentrations.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 Software. Student’s t test was used to compare data between two groups. To measure the strength of association between two variables, Pearson’s correlation was used. Values are expressed as mean values and standard deviations. Differences were regarded as statistically significant if P ≤ 0·05.
Results
Glutamine improved clindamycin-induced antibiotic-associated diarrhoea in mice
We first examined the effect of glutamine supplementation on AAD progression in a preclinical animal model. Oral administration of clindamycin for 2 weeks dramatically lowered body weight gain and increased faecal moisture, whereas it was attenuated by glutamine (Fig. 1(b) and (c)). Antibiotic exposure significantly lowered plasma and intestinal glutamine levels by 39 and 36 % (P < 0·001), while such effect could be partly normalised by glutamine supplementation.
Glutamine might be rapidly metabolised into other metabolites such as glutamic acid, arginine, asparagine and aspartic acid. In this regard, we noticed that arginine levels were elevated both in plasma and colonic tissues, whereas glutamic acid levels were significantly lowered upon clindamycin treatment (Fig. 1(d) and (e)).
The colonic glutamine depletion is generally due to increased metabolic demands beyond what can be met by endogenous synthesis or diet intake(Reference Ren, Ji and Tokar22). Although the expression of GS and SLC1A5 seemed to be affected by clindamycin (Fig. 1(f)), those findings should be carefully interpreted. Multiple genes (e.g., SLC1A5, SLC7A5, Gs, Gdh, Gpt2 and even c-Myc) have been implicated in glutamine metabolism, and it did remain a challenge to define molecular underlying mechanism.
Glutamine rectified clindamyci-induced intestinal barrier dysfunction
Derived from gut microbiota, LPS and d-lactate are regarded as biomarkers of intestinal permeability(Reference Shi, Kellingray and Zhai13,Reference Shi, Zhai and Li14) . As expected, plasma levels of LPS and d-lactate in clindamycin group were much higher than the control group (Fig. 2(a) and (b)). Consistent with the findings above, clindamycin affected tight junction protein and pro-inflammatory cytokine production in colonic tissues (Fig. 2(c)–(f)). Once again, such gut barrier dysfunction could be partially restored by glutamine supplementation.

Fig. 2. Glutamine restored clindamycin-induced intestinal barrier dysfunction. (a) Plasma d-lactate levels. (b) Plasma lipopolysaccharide (LPS) levels. (c) Colonic expression of tight junction protein occludin. (d) Colonic expression of tight junction protein zonula occludens 1 (ZO-1). (e) Colonic IL-6 levels. (f) Colonic TNF-α levels. Data are mean values and standard deviations (n 10 in each replicate). Significant difference compared with each respective vehicle group: ∗ P < 0·05, ** P < 0·01, *** P < 0·001.
Glutamine participated in M1 macrophage polarisation
Glutamine might serve as a fuel for colonic and immune cells. In the connection, we observed that the depletion of colonic glutamine greatly affected colonic epithelial cell turnover as well as wound healing (Fig. 3). We next questioned the potential influence of glutamine on macrophage polarisation both in primary peritoneal macrophages and RAW264.7 cells. Data clearly indicated that glutamine was essential for LPS-induced M1 polarisation. Mechanistically, glutamine might strengthen the intestinal antimicrobial barrier function via nitric oxide synthase 2 (NOS2)-dependent NO production (Fig. 4(a)–(c)). According to intracellular bacterial loading data, glutamine was also required for macrophage warding off microbial infection (Fig. 4(d)). It should be pointed out that glutamine did not affect E. coli growth in pure cultures. Taken together, glutamine participated in M1 macrophage polarisation.

Fig. 3. Importance of glutamine (Gln) in intestinal physical barrier function. (a) Glutamine is required for intestinal epithelial cell proliferation. HCT116 cell growth was measured by MTS assay as described in ‘Materials and methods’. Data are mean values and standard deviations (n 4 in each replicate). ![]() , 0 mm Gln;
, 0 mm Gln; ![]() , 2 mm Gln;
, 2 mm Gln; ![]() , 4 mm Gln. (b) Glutamine depletion delayed wound healing. Cell scratch assay data are presented as mean values and standard deviations from three independent experiments. *** Significant difference compared with the control group (P < 0·001).
, 4 mm Gln. (b) Glutamine depletion delayed wound healing. Cell scratch assay data are presented as mean values and standard deviations from three independent experiments. *** Significant difference compared with the control group (P < 0·001). ![]() , 24 h;
, 24 h; ![]() , 48 h. OD, optical density.
, 48 h. OD, optical density.
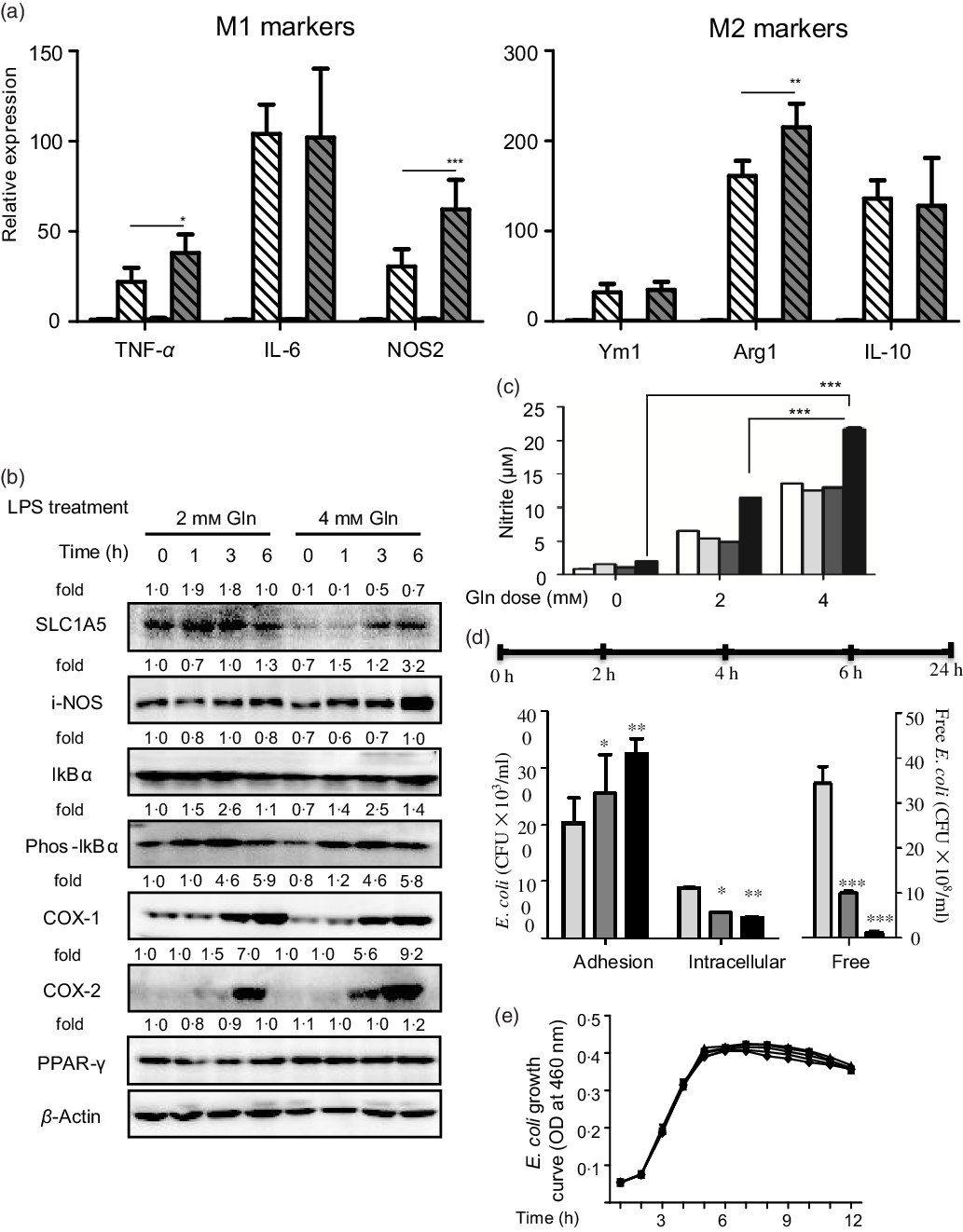
Fig. 4. Glutamine is critical for intestinal antimicrobial barrier function. (a) Effect of glutamine depletion on macrophage polarisation. Murine primary peritoneal macrophages were isolated and stimulated with lipopolysaccharides (LPS) or IL-4 as described in ‘Materials and methods’. Data are mean values and standard deviations (n 4 in each replicate). ![]() , 2 mM Gln;
, 2 mM Gln; ![]() , 2 mm Gln + LPS;
, 2 mm Gln + LPS; ![]() , 4 mm Gln;
, 4 mm Gln; ![]() , 4 mm Gln + LPS. (b) Effects of glutamine depletion on LPS-induced pro-inflammatory protein expression.
, 4 mm Gln + LPS. (b) Effects of glutamine depletion on LPS-induced pro-inflammatory protein expression. ![]() , 2 mM Gln;
, 2 mM Gln; ![]() , 2 mm Gln + IL-4;
, 2 mm Gln + IL-4; ![]() , 4 mm Gln;
, 4 mm Gln; ![]() , 4 mm Gln + IL-4. (c) NO release. Data are mean values and standard deviations (n 4 in each replicate).
, 4 mm Gln + IL-4. (c) NO release. Data are mean values and standard deviations (n 4 in each replicate). ![]() , 0 h;
, 0 h; ![]() , 1 h;
, 1 h; ![]() , 3 h;
, 3 h; ![]() , 6 h. (d) Effects of glutamine depletion on Escherichia coli infection in vitro. RAW264.7 cells were treated as described in ‘Materials and methods’. For E. coli infection, RAW264.7 cells were infected with E. coli, and intracellular bacterial load was assessed with a gentamicin protection assay.
, 6 h. (d) Effects of glutamine depletion on Escherichia coli infection in vitro. RAW264.7 cells were treated as described in ‘Materials and methods’. For E. coli infection, RAW264.7 cells were infected with E. coli, and intracellular bacterial load was assessed with a gentamicin protection assay. ![]() , 0 mm Gln;
, 0 mm Gln; ![]() , 2 mm Gln;
, 2 mm Gln; ![]() , 4 mm Gln. (e) Effects of glutamine on the growth of E. coli. Data are mean values and standard deviations (n 3 in each replicate).
, 4 mm Gln. (e) Effects of glutamine on the growth of E. coli. Data are mean values and standard deviations (n 3 in each replicate). ![]() , 0 mm Gln;
, 0 mm Gln; ![]() , 2 mm Gln;
, 2 mm Gln; ![]() , 4 mm Gln;
, 4 mm Gln; ![]() , 8 mm Gln;
, 8 mm Gln; ![]() , 16 mm Gln. NOS2, nitric oxide synthase 2; SLC1A5, solute carrier family 1 member 5; i-NOS, inducible nitric oxide synthase; COX-1, cyclo-oxygenase-1; COX-2, cyclo-oxygenase-2; Arg1, arginase-1; CFU, colony-forming units; OD, optical density. * P < 0·05, ** P < 0·01, *** P < 0·001.
, 16 mm Gln. NOS2, nitric oxide synthase 2; SLC1A5, solute carrier family 1 member 5; i-NOS, inducible nitric oxide synthase; COX-1, cyclo-oxygenase-1; COX-2, cyclo-oxygenase-2; Arg1, arginase-1; CFU, colony-forming units; OD, optical density. * P < 0·05, ** P < 0·01, *** P < 0·001.
Gut dysbiosis linked clindamycin exposure with glutamine metabolism
We eventually questioned how clindamycin lowered intestinal glutamine store. Gut microbiota is a novel environmental factor affecting energy harvest from the diet(Reference Backhed, Ding and Wang6) and even orchestrating the overall energy homoeostasis in host(Reference Chevalier, Stojanovic and Colin7). For colonic mucosa, butyrate from gut bacterial origin represents a major fuel under physiological conditions(Reference Lee and Hase8,Reference Cherbuy, Darcy-Vrillon and Morel9) . In this connection, we noticed that clindamycin exposure substantially confounded gut microbiota, evidenced by a decreased microbial diversity and a marked shift in community structure (Fig. 5(a) and (b)). Of note, the abundance of SCFA-producing bacteria strains (e.g. Bifidobacterium and Lactobacillus) as well as the faecal SCFA was greatly affected by clindamycin (Fig. 5(c)–(d)), whereas it could be partially rectified by glutamine supplementation.

Fig. 5. Effect of glutamine on clindamycin-induced gut dysbiosis. (a) Visualisation of principal coordinates (PC) analysis (PCoA) of unweighted UniFrac distances to show differences in bacterial community structure. ![]() , 1;
, 1; ![]() , 2;
, 2; ![]() , 3;
, 3; ![]() , 4;
, 4; ![]() , 5. (b) Heat map of differential taxa. Taxonomic heat map using Bray–Curtis dissimilarity index distance, combined with average (unweighted pair group method with arithmetic means) clustering for forty-one of the most statistically significant operational taxonomic units (OTU) between control and CTDA samples. Yellow and red represent high and low abundance, respectively. Class:
, 5. (b) Heat map of differential taxa. Taxonomic heat map using Bray–Curtis dissimilarity index distance, combined with average (unweighted pair group method with arithmetic means) clustering for forty-one of the most statistically significant operational taxonomic units (OTU) between control and CTDA samples. Yellow and red represent high and low abundance, respectively. Class: ![]() , Control;
, Control; ![]() , Glutamine;
, Glutamine; ![]() , Intervention;
, Intervention; ![]() , Model;
, Model; ![]() , Treatment. (c) Relative abundance of Bifidobacterium, Lactobacillus, Enterococcus and Enterobacter in faecal samples.
, Treatment. (c) Relative abundance of Bifidobacterium, Lactobacillus, Enterococcus and Enterobacter in faecal samples. ![]() , Proteus;
, Proteus; ![]() , Enterobacter;
, Enterobacter; ![]() , Sutterella;
, Sutterella; ![]() , Eubacterium;
, Eubacterium; ![]() , Coprobacillus;
, Coprobacillus; ![]() , Allobaculum;
, Allobaculum; ![]() , Oscillospira;
, Oscillospira; ![]() , Ruminococcus;
, Ruminococcus; ![]() , Coprococcus;
, Coprococcus; ![]() , Lactobacillus;
, Lactobacillus; ![]() , Enterococcus;
, Enterococcus; ![]() , Enterococcaceae;
, Enterococcaceae; ![]() , Prevotella;
, Prevotella; ![]() , Bacteroides;
, Bacteroides; ![]() , Bifidobacterium. (d) Faecal SCFA content. The contents of SCFA in mice faeces were measured by GC analysis. Data are mean values and standard deviations (n 10 in each replicate).
, Bifidobacterium. (d) Faecal SCFA content. The contents of SCFA in mice faeces were measured by GC analysis. Data are mean values and standard deviations (n 10 in each replicate). ![]() , Isovaleric acid;
, Isovaleric acid; ![]() , isobutyric acid;
, isobutyric acid; ![]() , butyric acid;
, butyric acid; ![]() , propionic acid;
, propionic acid; ![]() , acetic acid.
, acetic acid.
Discussion
In the present study, we demonstrated that long-term antibiotic use lowered colonic glutamine store, which in turn might increase the risk of opportunistic diarrhoeal pathogen infection. Considering its critical roles in physical and immunological barriers, glutamine store might serve as an ancient mechanism against microbial infection. Collectively, our findings in the present study might provide a rationale for guiding the clinical uses of glutamine in AAD management in future.
Gut dysbiosis has been implicated in the aetiology of AAD(Reference Dethlefsen and Relman23). Resilience to external perturbations is a fundamental property of gut microbial communities, and gut microbiome is generally stable within healthy adult individuals(Reference Sanders, Guarner and Guerrant24). However, antibiotic therapy might represent a repeated violent disturbance and often lead to a persistent regimen shift which is difficult to recover soon(Reference Dethlefsen and Relman23). To this end, probiotic has been proposed for AAD management. Although certain probiotic showed some promise, their overall efficacy is far to meet the standard of medical care(Reference Sanders, Guarner and Guerrant24). It should be pointed out that probiotic was not suit for critically ill hospitalised patients. An explanation for this issue is immune tolerance. Probiotic represents external micro-organism for host and thus might cause excessive immunity response, especially in patients with gastrointestinal disorders. Based on the findings in our study, one therapeutic approach to AAD management might be firstly to restore the intestinal mucosal barrier with glutamine and then to sustain remission by probiotic interventions(Reference Coeffier, Dechelotte and Ducrotte25).
More recently, gut microbiota has been implicated in host energy homoeostasis(Reference Finnie, Dwarakanath and Taylor26). It is not at all a surprise to find that antibiotic might disrupt host energy balance via gut microbiota. Butyrate from gut bacterial origin represents a major fuel for colonic mucosa under physiological conditions(Reference Lee and Hase8,Reference Cherbuy, Darcy-Vrillon and Morel9) . In previous germfree mice study, colonic epithelial cells might compensate the absence of butyrate by increasing glutamine use(Reference Cherbuy, Darcy-Vrillon and Morel9). In our study, clindamycin exposure changed gut microbiota composition especially in butyrate-producing strains and led to faecal butyrate deficiency. We wonder that it might in turn increase the body’s demand for glutamine. If it is a case, glutamine depletion might be a secondary event of gut dysbiosis. People might also question can SCFA rescue the Gln deficiency in AAD? SCFA are weak acids and their transport is mainly through monocarboxylate transporters, while those transporters were widely expressed in gastrointestinal tract(Reference Payen, Mina and Van Hee27). Thus, to address the issue above might be upon delivery methods, as SCFA might be rapidly absorbed by the upper gastrointestinal tract and most likely fail to reach the colon.
Although our study is intriguing, several questions remain unanswered. The first issue is the possibility of bench-to-bedside translation. For example, a major caveat in our mouse model is that loose stools, but not severe acute diarrhoea, were observed. One explanation is the difference in hygienic sanitary conditions between animal facility and hospital. On the other hand, glutamine was reported to mediate the expression of virulent factors in pathogens(Reference Si, Yuan and Chang28), and whether its supplementation might interfere with antibiotic efficacy remains unknown. Moreover, glutamine metabolism is often reported to be enhanced in tumours as well as precancerous lesions(Reference Ren, Ji and Tokar22). Taken together, the efficacy and safety of glutamine supplementation should be rigorously studied by randomised, double-blind, placebo-controlled trials. Another issue is that the mechanism underlying glutamine supplementation against AAD remains unclear. The direct evidence between glutamine and gut microbiota dysbiosis was not available yet. Faecal microbiota transplantation from glutamine-treated donors to mice suffering AAD might partly address this issue(Reference Wu, Esteve and Tremaroli19). In addition to classical antimicrobial macrophage activation, we also examined the potential influence of glutamine on M2 polarisation. Among IL-4-mediated M2 marker genes, glutamine greatly enhanced arginase-1 expression (Fig. 4(a)). Considering the importance of arginase-1 polyamine axis in wound healing(Reference Bachmann, Leonard and Delzenne29), we reasoned that glutamine metabolism might function in either initiation or resolution of acute inflammatory response. To gain deeper insight into the role of glutamine metabolism in intestinal mucosal barrier integrity, further study examining colonocyte epithetical cell turnover and tight junction protein expression as well as distribution is needed. Most likely, glutamine might act through diverse mechanisms to maintain gut health.
In summary, our study revealed that glutamine might become a ‘conditionally essential’ amino acid in patients receiving long-term antibiotic therapy, and its supplementation might hold promise for AAD management.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81773064, 31972973 and 32021005), National Youth 1000 Talents Plan, the Jiangsu Specially-Appointed Professor Program, Jiangsu Province Recruitment Plan for High-level, Innovative and Entrepreneurial Talents (Innovative Research Team), and Collaborative Innovation Center of Food Safety and Quality Control in Jiangsu Province.
W. C., H. L. and H. Z. designed and supervised the experiments. J. M. and H. L. prepared the manuscript. J. M., Y. Y., H. L. and X. S. performed experiments.
No potential conflicts of interest were disclosed.
Supplementary material
For supplementary material referred to in this article, please visit https://doi.org/10.1017/S0007114520004195







