


More than half a billion people are obese worldwide, causing an estimated 2·8 million deaths each year due to metabolic comorbidities, such as CVD and type 2 diabetes (T2D)( 1 ). Metagenomic studies have suggested that obesity-related metabolic diseases are accompanied by alterations in gut bacteria gene composition and abundance( Reference Turnbaugh, Hamady and Yatsunenko 2 – Reference Karlsson, Tremaroli and Nookaew 7 ). Noticeably, microbial genes seem to be a stronger predictor of T2D than common anthropometric risk markers( Reference Karlsson, Tremaroli and Nookaew 7 ) and variation in the human genome( Reference Qin, Li and Cai 3 ), as shown in European and Chinese individuals, respectively, although the most discriminatory microbial genes differed between the Chinese( Reference Qin, Li and Cai 3 ) and European individuals( Reference Karlsson, Tremaroli and Nookaew 7 ).
Still, it is not clear whether shifts in the gut microbiota can cause metabolic diseases, or if they are just a consequence. However, causality is suggested by studies in germ-free mice( Reference Backhed, Ding and Wang 8 , Reference Turnbaugh, Ley and Mahowald 9 ), and one study in human subjects( Reference Vrieze, Van Nood and Holleman 10 ). These studies have shown that transplantation of gut microbiota can induce modifications in the microbiota of the receiving host, associated with either impaired or improved metabolic health, depending on the phenotype of the donor. Studies in human subjects that have measured diet-induced alterations in gut metagenomic and whole-body metabolic markers simultaneously have shown that modifications in the microbiota are accompanied by improvements in glucose homeostasis and lipid metabolism( Reference Dewulf, Cani and Claus 11 , Reference Vulevic, Juric and Tzortzis 12 ). Together, these studies suggest that the gut microbiota constitute a promising target in the prevention of metabolic diseases.
Food components that directly target the gut microbiota include pre- and probiotics. Probiotics are living micro-organisms that, when ingested, provide health benefits, either directly through interactions with host cells or indirectly through effects on other bacterial species( Reference Gordon 13 ). Common probiotics include Lactobacillus species( Reference de Vrese and Schrezenmeir 14 ), and Lactobacillus paracasei has previously been associated with a healthy metabolic profile( Reference Million, Maraninchi and Henry 15 , Reference Stsepetova, Sepp and Kolk 16 ). Prebiotics are dietary fibres that are selectively fermented by the gut bacteria and induce specific changes in the composition and/or activity of the gut microbiota that provide benefits to host health( Reference Gibson, Scott and Rastall 17 ). Flaxseed (Linum usitatissimum L.) contains approximately 30 % of dietary fibres, of which one-third are soluble viscous fibres (mucilage)( Reference Hall, Tulbek and Xu 18 ) that induce beneficial effects on glucose homeostasis and lipid metabolism in human subjects( Reference Cunnane, Ganguli and Menard 19 – Reference Kristensen, Jensen and Aarestrup 21 ). Flaxseed fibres have been shown to be highly fermentable in rats( Reference Berggren, Björck and Nyman 22 ); however, it is not known whether beneficial metabolic effects of flaxseed mucilage on human subjects can be explained by modulation of the gut microbiota.
The aim of the present study was to explore the effect of dietary interventions either with L. paracasei F19 or with flaxseed mucilage on gut microbiota and metabolic risk markers in obese postmenopausal women.
Materials and methods
A total of fifty-eight women were randomised to a parallel-group intervention of 6 weeks' duration with a daily intake of L. paracasei ssp. paracasei F19, flaxseed mucilage or placebo (Fig. 1). The participants were blinded to their allocation; L. paracasei F19 was mixed with maltodextrin and administered in sachets and the indistinguishable placebo product was administered in sachets with a corresponding dose of maltodextrin. Flaxseed mucilage was administered in breakfast buns and the corresponding placebo products were buns without flaxseed mucilage.

Fig. 1 Illustration of the 6-week parallel-group intervention, with participants randomised to one of the three different diet groups.
The primary outcomes were the effect on the gut microbiota composition and insulin sensitivity; the secondary outcomes were the effect on the inflammatory markers, blood lipids and fat mass distribution. Enrolment of participants was performed continuously by the same two researchers, and randomisation was carried out continuously using pre-prepared opaque sealed envelopes with an equal allocation ratio of 1:1:1. Sample size calculations were based on preliminary results from a subgroup of Danish participants in the Metagenomics of the Human Intestinal Tract (MetaHIT) study( Reference Le Chatelier, Nielsen and Qin 5 ). With a sample size of eighteen participants per group, a statistical power of 80 %, a two-sided significance level of 0·05 and with the assumption of equal variances, the study will identify the changes between independent groups in gut microbiota composition corresponding to 1 sd, e.g. a difference of 40 000 gene counts will be identified if the sd is 40 000 gene counts, and similarly, a 0·5 difference in insulin sensitivity (homeostatic model assessment of insulin resistance, HOMA-IR) will be identified for an sd of 0·5.
The participants attended a screening visit within the 2 weeks before randomisation and four visits after enrolment: visit 1 at baseline (day 0); visit 2 at day 28 of the intervention; visit 3 at the end of the intervention (day 42) and visit 4 4 weeks after the end of the intervention. After the completion of the study, the participants were offered a 10-week weight reduction programme. During the visits, blood was drawn, faecal samples were collected, body composition and blood pressure were measured, and information on dietary intake, physical activity level and adverse events were obtained. The present analyses included the 6-week intervention period that was the primary outcome of the study.
Study population
Participants were recruited from the Copenhagen area through advertisements in newspapers and relevant web sites. The inclusion criteria were women aged 40–70 years, ≥ 1 year since last menstruation, BMI of 30–45 kg/m2, waist circumference >80 cm and leucocyte blood count >4·7 × 109 cells/l. The exclusion criteria were gastrointestinal diseases, chronic diseases (such as type 1 diabetes or liver cirrhosis), medically treated T2D or dyslipidaemia, intake of antibiotics within the previous 3 months, intake of supplementary pro- or prebiotics or abnormal high quantities of fermented foods (>400 g/d) in the previous 6 weeks, or inability to comply with the research protocol. The present study took place at the Department of Nutrition, Exercise and Sports, Faculty of Sciences, University of Copenhagen, Denmark, from September 2011 to September 2012.
The present study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human participants were approved by the Ethics Committee for the Capital Region of Denmark (journal H-3-2011-067). Written informed consent was obtained from all participants. The present study was registered at ClinicalTrial.gov (NCT01433120).
Intervention
The probiotic product contained L. paracasei F19 (9·4 × 1010 colony-forming units/dose). The probiotic and the placebo (pure maltodextrin) sachets were indistinguishable. The products were stored at − 80°C until the weekly supply to the participants, after which they were stored in their personal freezer at − 18°C until consumption. Participants dissolved the entire content of the sachet in a glass of water and consumed it in the morning. Bacterial viability was confirmed at the end of the intervention by anaerobic culture tests of a sample of the products. Dilution of samples was spread on deMan, Rogosa and Sharpe (MRS) pH 5·4 agar. Plates were incubated anaerobically 72 h at 37°C. All characteristic colonies were checked by microscopy and counted (Arla Foods amba).
Flaxseed mucilage was extracted from whole flaxseed under heat treatment (Biogin Biochemicals Company Limited). Breakfast buns based on wheat with and without flaxseed mucilage were produced by the Department's experimental kitchen, and the daily amount of flaxseed mucilage administered to the active group was 10 g (5 g/bun). The products were stored at − 18°C at the Department until the weekly supply to the participants. The intervention and placebo buns were comparable in visual appearance, taste and macronutrient composition. They were consumed during the morning hours. The participants were otherwise instructed to maintain habitual dietary habits.
Participants were required to keep a diary of their intake of study products, and these were collected at the weekly visits at the Department. A compliance score was calculated as the percentage of the prescribed test products a participant reported to have consumed during the 6 weeks of intervention. The participants were interviewed about all types of potential adverse effects at each visit by the use of broad, open-ended questions; in addition, they were asked specifically to changes in stool characteristics (consistency and frequency).
Body composition, dietary and activity records
Body composition was assessed by dual-energy X-ray absorptiometry (iDXA; Lunar Radiation Company), BMI (kg/m2), waist circumference and intra-abdominal adipose tissue (cm2: − 208·2+4·62 (sagittal diameter, cm)+0·75 (age, years)+1·73 (waist, cm)+(0·78 (trunk fat, %)))( Reference Treuth, Hunter and Kekes-Szabo 23 ). Registration of 3 d weighed dietary intake and physical activity level were performed within the week before each visit. A registered dietitian analysed all dietary records using the Danish dietary software program (Dankost Pro). Physical activity was registered using the Physical Activity Scale( Reference Aadahl and Jorgensen 24 ) where the metabolic equivalent value is calculated based on the time spent on nine different intensity levels ranging from sleep to very strenuous activities during 24 h.
Biochemical analyses
Fasting blood samples were obtained after an overnight fast followed by an oral glucose tolerance test (OGTT) where 75 g of glucose were dissolved in 300 ml of water and consumed within 5 min. During the OGTT, blood samples were taken with 30 min intervals for 3 h to ensure return to baseline values. Blood for glucose analysis was collected in iced sodium fluoride tubes. Blood samples for the analysis of leucocytes count, high-sensitivity C-reactive protein (hsCRP), lipopolysaccharide-binding protein (LBP), IL-6 and angiopoietin-like protein 4 (ANGPTL4) were drawn in iced EDTA tubes. Blood samples for all other analyses were collected in non-coated tubes. Blood samples were centrifuged for 10 min at 2500 g at 4°C and kept at − 80°C until analyses were performed. Insulin resistance was assessed by glucose, insulin and C-peptide at the fasting and stimulated state. Inflammatory state was assessed by leucocytes count, hsCRP, TNF-α, IL6 and LBP. Lipid metabolism was assessed by measurement of serum concentrations of total cholesterol, LDL-cholesterol, HDL-cholesterol and TAG, and plasma concentration of ANGPTL4. ABX Pentra 400 (Horiba ABX) was used to analyse glucose (intra- and inter-assay CV: 1·1 and 1·5 %), TAG (intra- and inter-assay CV: 3·8 and 3·0 %), total cholesterol (intra- and inter-assay CV: 1·0 and 1·7 %), HDL-cholesterol (intra- and inter-assay CV: 1·2 and 2·7 %), LDL-cholesterol (intra- and inter-assay CV: 1·3 and 3·3 %) and hsCRP (intra- and inter-assay CV: 3·6 and 6·5 %). Immulite 1000 (Siemens Medical Solutions Diagnostics) was used to analyse insulin (intra- and inter-assay CV: 4·2 and 7·3 %) and C-peptide (intra- and inter-assay CV: 6·9 and 5·2 %). Sysmex KX-21 hematology (Sysmex GmbH) was used to analyse the leucocytes count (intra- and inter-assay CV: 2·0 and 1·4 %). LBP (intra- and inter-assay CV: 6·1 and 9·8–17·8 %) was analysed with a human LBP Elisa kit (Abnova). TNF-α (intra- and inter-assay CV: 5·4 and 6·1 %), IL-6 (intra- and inter-assay CV: 7·4 and 8·5 %) and ANGPTL4 (intra-assay CV: 3·8 %) were analysed with ELISA (antibodies were purchased from R&D Systems). Insulin resistance was estimated by HOMA-IR( Reference Matthews, Hosker and Rudenski 25 ). Data from the OGTT were evaluated by AUC analysis and by the Matsuda index( Reference Matsuda and DeFronzo 26 ).
Stool samples and microbiota analyses
Participants collected samples of faeces in two 20 ml tubes (samples A and B) within 2 d before the visit. The samples were either stored immediately at − 80°C or briefly stored in personal − 18°C freezers before transport to the laboratory in cooled containers. Total faecal DNA from the fifty-three participants who completed the dietary intervention was extracted, sequenced and analysed by quantitative metagenomics at Metagenopolis (INRA). To obtain homogenous representation of all bacterial species, DNA extraction involved use of quenching solutions that protect DNA potentially prematurely released by lysis of fragile cells from degradation by DNases present in the stools and a bead-beating step that assures lysis of particularly robust cells( Reference Suau, Bonnet and Sutren 27 , Reference Godon, Zumstein and Dabert 28 ). A Barcoded Fragment Library was prepared for each sample and DNA sequencing data were generated using the SOLiD 5500xl sequencers (Life Technologies). An average of 65 (sd 48) million 50-base-long single reads was determined for each sample.
Primary data analyses, from reads to generation of a raw count matrix, were performed using METEOR Studio pipeline for quantitative metagenomic profiling developed at INRA MetaGenoPolis based on the iMOMi database. Reads generated from the SOLiD sequencer were trimmed to thirty-five bases and then mapped on the reference catalogue of 3·3 million genes( Reference Qin, Li and Raes 29 ) using Bowtie software with a maximum of three mismatches and selection of the best hit. If multiple alignments were found, counts were divided equally between the aligned genes. Using both thirty-five bases reads and three maximum mismatches allows to take into account the strain variability and the non-redundant nature of the gene catalogue where redundant open reading frames were removed using a criterion of 95 % identity over 90 % of the shorter open reading frame length. Average of 38 (sd 14) million reads per sample was thus mapped and used to construct a raw gene count matrix.
Secondary analyses, from matrix normalisation to microbiota analyses, were performed using MetaOMineR package, an analytical suite (R language) developed at the INRA MetaGenoPolis( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ) that contains different algorithms and routines to normalise and analyse raw gene count matrixes to extract biological signals. To decrease technical bias due to different sequencing depth, 24 million reads were randomly selected for each sample using a draw without replacement. Abundance of each gene in a sample was normalised by dividing the number of reads that uniquely mapped to a gene by its nucleotide length. Then, normalised gene abundance was transformed in to frequencies by dividing with 24 million (see online Supplementary Fig. S1). The resulting set of gene frequencies, the microbial gene profile of an individual, was used for further analyses.
Microbial gene richness was measured by counting the number of genes that are present for a given sample using a downsized count matrix as performed in the original studies( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ). To be able to compare the present study and the former ones, we used the same method, estimating gene count richness using at 11 million unique reads matrix and a gene richness categorical variable computed by applying a threshold of 480 000 bacterial genes, to distinguish low from high gene count( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ). A prediction model for bacterial richness based on the six bacterial species was applied in a receiver operating characteristic analysis as described previously( Reference Le Chatelier, Nielsen and Qin 5 ), computing the sum of mean abundance of species with greater abundance in high gene count than in low gene count minus the sum of those with greater abundance in low gene count than in high gene count. Richness was estimated using either shared or unique matrix downsized matrices, and either gene count for richness or richness index such as exponential of Shannon richness index or inverse Simpson index.
Differentially abundant genes between both time points were selected for each diet using paired Wilcoxon test (P< 0·01), then clustered into metagenomic species (MGS), using the method based on binning co-abundant genes across all individuals samples reported previously( Reference Le Chatelier, Nielsen and Qin 5 ). To verify that the genes from a given cluster belonged to the same genome and to annotate the MGS taxonomically, we performed blastN and blastP analyses using a collection of 6006 genomes (the available reference genomes from the National Center for Biotechnology Information and the set of draft gastrointestinal genomes from the Data Analysis and Coordination Center of the HMP and MetaHIT (3 August 2012 version)). MGS were assigned to a given genome when more than 80 % of the genes matched the same genome using blastN, at a threshold of 95 % identity over 90 % of gene length. The remaining MGS were annotated using blastP analysis and assigned to a given taxonomical level from genus to super kingdom level if more than 80 % of their genes had the same level of assignment. Data were indexed in a relational database permitting downstream access to genes, gene function, phylogeny and covariation analysis.
Quantification of SCFA using the ethyl chloroformate-NEFA method
For the quantification of SCFA, approximately 10 g of stool were mixed with an equal amount of deionised H2O (18·2 MΩ) filtered water (0·22 mm). Sample preparation, analyses by the ethyl chloroformate NEFA method and GC, were performed by applying methods described in detail by Amer et al. ( Reference Amer, Nebel and Bertram 30 ). Quantification of ethyl chloroformate-NEFA esters was obtained using external calibration curves for each NEFA. The two isotopic standards butyric internal standard (D7, 98 %) and Cambridge Isotope Laboratories, Inc. were used as internal standard for butyric acid.
Statistical analyses
Effect of dietary interventions on biochemical and anthropometric parameters was analysed for completers by multiple linear regression models adjusted for baseline values, with stepwise backward elimination of the following covariates: age; body fat percentage (baseline); energy intake in the week preceding the last visit; changes in weight and physical activity level during the intervention. Log-transformation was applied to non-normally distributed variables. Comparisons of two means were performed using t tests. Regression coefficients, CI and P-values are reported from the adjusted regression models, while non-adjusted mean values for changes in biochemical parameters during the intervention are reported in Table 2. Correlation analyses between bacterial gene count and biochemical measures were analysed by Spearman's rank correlation coefficient. At an explorative level, the potential effect of diet-induced compositional changes in the gut microbiota on metabolic markers was examined by separate analyses where MGS that changed during the intervention were included as explanatory variables in the multiple regression models. Statistical significance was considered at a threshold value of 0·05. Analyses were performed using MetaOMinerR package (developed at Metagenopolis, INRA) and JMP version 9.0.2 (SAS Institute, Inc.).
Results
A total of fifty-three participants completed the study (91 %). Dropout rates were not significantly different between groups, and no significant differences were found between dropouts and completers in anthropometric and biochemical characteristics at baseline. Details on recruitment, randomisation and study flow are shown in Fig. 2. Baseline characteristics for all the participants by intervention group are presented in Table 1. There were no differences in the anthropometric measures between groups; however, the participants in the L. paracasei F19 group had significantly impaired insulin sensitivity compared with the flaxseed mucilage group, and a significantly lower bacterial gene count compared with the placebo group. All the participants reported to have consumed at least 75 % of the study products during the 6 weeks of intervention, and the mean compliance score was above 90 % in all the groups.

Fig. 2 Flow chart of the study.
Table 1 Baseline characteristics presented by diet group (Mean values and standard deviations)

IAAT, intra-abdominal adipose tissue; E%, energy percentage; 24-h-MET-time, 24-h metabolic equivalent value; HOMA-IR, homeostatic model assessment of insulin resistance; ANGPTL4, angiopoietin-like protein 4; hsCRP, high-sensitivity C-reactive protein; LBP, lipopolysaccharide-binding protein.
* The analyses are only performed in fifty-three completers.
Diet-induced changes in biochemical markers
Multiple linear regression analyses of the effect of dietary intervention on biochemical markers showed that the intake of flaxseed mucilage over 6 weeks improved insulin sensitivity as shown by a decrease in the serum C-peptide and insulin response following the OGTT. The AUC values for serum C-peptide and insulin were 337 pmol/l per min (95 % CI 168·7, 505·1) and 65·9 pmol/l per min (95 % CI 26·5, 105·2) lower compared with placebo (adjusted values), corresponding to 12 and 13 % improvement within the flaxseed mucilage group (Table 2). Matsuda's index was increased by 0·4 (95 % CI 0·1, 0·7), compared with placebo (adjusted values); corresponding to 11 % increase within the flaxseed mucilage group. Markers for lipid metabolism and inflammation were reduced after 6 weeks in the flaxseed group (Table 2); however, there were no significant differences compared with placebo. There was no effect of L. paracasei F19 on insulin sensitivity, lipid metabolism, inflammatory markers or anthropometric measures compared with placebo (Table 2).
Table 2 Biochemical characteristics after 6 weeks of dietary intervention and changes from baseline (Mean values with their standard errors)

Δ, change during the 6 weeks of dietary intervention (unadjusted values); HOMA-IR, homeostatic model assessment of insulin resistance; ANGPTL4, angiopoietin-like protein 4; hsCRP, high-sensitivity C-reactive protein; LBP, lipopolysaccharide-binding protein.
* P-values are reported from linear regression models of the effect of dietary group on the specified parameters, when adjusted for relevant covariates including baseline values.
† The difference is to be found between the F19 and flaxseed mucilage group, but not when these are compared with placebo by Student's t test.
Diet-induced changes in gut microbiota composition
Sequenced reads mapped against the MetaHIT gene catalogue of 3·3 million genes at a normal range of 61 (se 6) %. Of the fifty-three participants, forty-nine (92·5 %) had high gene count and four (7·5 %) had low gene count; two in the group allocated to L. paracasei F19 and two in the group allocated to flaxseed mucilage. A prediction model for bacterial richness based on six bacterial species, introduced by Cotillard et al. ( Reference Cotillard, Kennedy and Kong 4 ), was applied to the current data set and an AUC value of 0·94 was obtained.
At baseline, total bacterial gene count, which is a measure of gut bacterial richness, correlated negatively with total cholesterol (r − 0·28, P= 0·043), LDL-cholesterol (r − 0·30, P= 0·029), leucocytes count (r − 0·34, P= 0·015) and aspartate aminotransferase (r − 0·33, P= 0·018), and tended to correlate negatively with AUC glucose (r − 0·25, P= 0·069) and alanine aminotransferase (r − 0·24, P= 0·091).
Within the flaxseed mucilage group, bacterial gene count decreased with 47 271 genes over 6 weeks (95 % CI − 82 176, − 12 366, P= 0·011); however, this change was not significantly compared with placebo (P= 0·144). Bacterial gene count did not change within the L. paracasei F19 group (P= 0·473). The gene loss following intake of flaxseed mucilage for 6 weeks was confirmed by measures of α diversity; when evaluated by the exponential of Shannon diversity index and Simpson's inverse index, the decrease in diversity was 38 010 (95 % CI − 64 473, − 11 546, P= 0·007) and 17 515 (95 % CI − 30 992, − 4038, P= 0·014), respectively.
Comparison of microbiota composition at baseline and after 6 weeks of intervention with L. paracasei F19 showed alterations in faecal abundance of 2493 bacterial genes assigned to two MGS that increased during the intervention relative to baseline. These were identified as the species Eubacterium rectale and Ruminococcus torques. The proportion of total distributions of E. rectale recovered after analysis increased 3·3 times (P= 0·003) and R. torques increased 4·5 times (P< 0·001).
The placebo intervention led to altered faecal abundance of 7436 genes assigned to six MGS (P< 0·01), where the relative abundance of four MGS (Roseburia hominis, two Clostridiales and one unknown) decreased and the relative abundance of two MGS (Eubacterium ventriosum and one unknown) increased.
Comparison of microbiota composition at baseline and after 6 weeks of intervention with flaxseed mucilage showed alterations in faecal abundance of 41 090 bacterial genes assigned to thirty-three MGS, the relative abundance of twenty-four MGS decreased and nine MGS increased (Table 3 and Fig. 3). Of the thirty-three MGS that changed, twenty-four were identified at phylum level or below. Eight of the MGS identified as decreasing during the intervention were assigned to the Faecalibacterium genus, among which three F. prausnitzii species with sequence homology to the strains F. prausnitzii A2-165, F. prausnitzii SL3/3 and F. cf. prausnitzii KLE1255. The proportion of total distributions of these three species decreased 0·6 times (P< 0·01). Moreover, Ruminococcus lactaris decreased 0·9 times during the flaxseed mucilage intervention (P= 0·005).
Table 3 Gut bacteria changes in relative abundance after 6 weeks of intervention with 10 g flaxseed mucilage
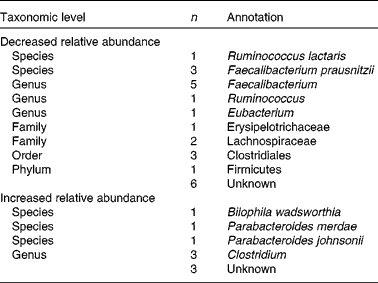

Fig. 3 The presence and abundance of the thirty-three species that differed significantly before and after 6 weeks of intervention with flaxseed mucilage. Each barcode illustrates the abundance of a species, either enriched at baseline (top) or week 6 (bottom): samples are in columns (same order for both time points) and the fifty ‘tracer’ genes are in rows. Gene abundance is indicated by colour gradient from white (not detected) over blue, green and yellow to red (most abundant). Taxonomical information is given for each species; the number of genes within each metagenomic species is given in parenthesis. Fdr, Benjamini Hochberg multiple testing correction of paired Wilcoxon tests; unk., unknown taxonomy.
Three of the nine MGS that increased in relative abundance after the flaxseed mucilage intervention were assigned to the Clostridium genus (Table 3 and Fig. 3). Furthermore, three MGS were identified to species level as Bilophila wadsworthia, Parabacteroides merdae and Parabacteroides johnsonii. The proportion of total distributions of these three species increased 2·6 times (P< 0·001), 3·6 times (P= 0·004) and 4·7 times (P< 0·001), respectively.
Effect of gut microbiota on the improvement in insulin sensitivity
When exploring the role of relative changes in the gut microbiota on improvement in insulin sensitivity in the flaxseed mucilage group, the MGS that changed during the intervention were included in the multiple regression models in separate analyses. Relative changes in the MGS did not contribute to the effect of the flaxseed mucilage intervention on Matsuda index or AUC values for insulin and C-peptide, when separate analyses were performed where the delta values for the individual MGS were included in the model as explanatory variables.
Adverse events
During the initial 4 weeks of the intervention, more adverse events were reported in the flaxseed group (n 11), compared with the placebo group (n 4; P= 0·023), while there was no difference in the occurrence of adverse events between the placebo and the L. paracasei F19 group (n 8; P= 0·297). The adverse events reported following the flaxseed intervention were increased flatulence and changed bowel habits with more frequent defecation and looser stool consistency. After 6 weeks, these symptoms were diminishing and only reported in seven participants, which was not different from the placebo group. There was no difference between the adverse events reported following the probiotic and placebo intervention that included both more frequent and less-frequent defecation. No serious adverse events were registered during the study.
Discussion
The present study shows that daily intake of flaxseed mucilage over 6 weeks can improve insulin sensitivity and modify the gut microbiota in individuals with obesity. The relative decrease in the abundance of F. prausnitzii species following intake of flaxseed mucilage is surprising given the improvement in insulin sensitivity. Previous metagenomic studies have suggested that F. prausnitzii is more abundant in healthy individuals than in individuals with T2D( Reference Qin, Li and Cai 3 , Reference Karlsson, Tremaroli and Nookaew 7 ). In addition, prebiotics have been shown to induce a parallel increase in the faecal abundance of F. prausnitzii and improvement in insulin sensitivity in individuals with obesity( Reference Dewulf, Cani and Claus 11 ). A beneficial effect of F. prausnitzii on insulin sensitivity has been hypothesised to be due to its role as a major producer of the SCFA butyrate( Reference Louis and Flint 31 ), as butyrate seems to have an anti-inflammatory potential that might improve obesity-related metabolic complications such as insulin resistance( Reference Brahe, Astrup and Larsen 32 ). The relative lower abundance of F. prausnitzii following intake of flaxseed mucilage is supported by the reduction observed in faecal butyrate content and by results from a previous study performed in rats, where fermentation of flaxseed fibres had been shown to yield a remarkably low proportion of butyrate, when compared with other dietary fibres( Reference Berggren, Björck and Nyman 22 ).
The improvement in insulin sensitivity following intake of flaxseed mucilage was accompanied by reductions in markers for inflammation and dyslipidaemia, although these changes were non-significant, compared with the placebo group. The gut microbiota has been implicated in the low-grade inflammation that characterises the progression from obesity to metabolic disease, as translocation of bacterial toxins, such as lipopolysaccharides, into the circulation can activate inflammatory pathways( Reference Sun, Yu and Ye 33 , Reference Pussinen, Havulinna and Lehto 34 ). Impaired gut barrier function can be caused by adipocyte-derived inflammatory cytokines( Reference de Kort, Keszthelyi and Masclee 35 ), and increased translocation of lipopolysaccharides can be induced by a high-fat diet( Reference Erridge, Attina and Spickett 36 , Reference Ghoshal, Witta and Zhong 37 ). Thus, it is likely that the gut is an important mediator in obesity-related systemic inflammation. Prebiotic-induced modulation of the gut microbiota has been proposed to inhibit translocation of lipopolysaccharides based on observations in mice( Reference Cani, Neyrinck and Fava 38 ); however, it is still unclear to what degree such mechanisms apply to human subjects. Since the decrease in LBP, hsCRP, TNF-α and IL-6 in the present study did not differ between the flaxseed mucilage and the placebo group, the improvement in insulin sensitivity did not appear to be mediated through inhibition of lipopolysaccharide translocation.
The design of the present study allowed us to explore whether the improvement in insulin sensitivity following intake of flaxseed mucilage could be due to a gut microbiota modulating effect. However, analyses showed that the changes in relative abundance of bacterial species induced by flaxseed mucilage did not explain any of the changes in the biochemical markers. When this is considered in relation to the flaxseed mucilage-induced changes in the microbiota that, based on the existing literature, are not in line with an improved host metabolic phenotype, we conclude that changes in the gut microbiota following intake of flaxseed mucilage do not appear to be a contributing factor to the beneficial effect of flaxseed mucilage on insulin sensitivity. Previously, it has been shown that daily intake of 5 g flaxseed mucilage reduced fasting blood glucose, total cholesterol and LDL-cholesterol in patients with T2D after 12 weeks( Reference Thakur, Mitra and Pal 20 ), and reduced total- and LDL-cholesterol in healthy individuals after 1 week( Reference Kristensen, Jensen and Aarestrup 21 ). In addition, acute meal tests with flaxseed mucilage have been shown to suppress postprandial blood glucose, insulin and lipid responses( Reference Cunnane, Ganguli and Menard 19 , Reference Kristensen, Savorani and Christensen 39 ). The beneficial metabolic effects of flaxseed mucilage observed in these previous studies and in the present study would then appear to be due to the ability of the soluble viscous fibres to delay gastric emptying and inhibit nutrient absorption rather than their ability to induce specific changes in the gut microbiota. Although, it cannot be excluded that the effect on insulin sensitivity was mediated through undetected bacterial species or changes in proteins and metabolites present in the gut that were not measured in the present study. The dominating molecular pathways in the bacterial species that increased during the flaxseed mucilage intervention are involved in pro-inflammatory signalling and managing of oxidative stress, pathways that have previously been suggested to be enriched in individuals with obesity-related metabolic diseases( Reference Le Chatelier, Nielsen and Qin 5 ). This shift towards a gut microbiome with a more pro-inflammatory potential might be explained by the fact that the flaxseed mucilage intervention provided more Cd in the diet. Despite that the Cd content in the flaxseed mucilage was at a concentration considered harmless to human health (0·113 mg/kg)( Reference Tarpila, Wennberg and Tarpila 40 ), it still might have induced modifications in the gut microbiome. This has been observed in previous studies in mice, where alterations were induced in the gut microbiome following administration of environmental relevant low doses of Cd in drinking-water over 3–8 weeks( Reference Fazeli, Hassanzadeh and Alaei 41 – Reference Liu, Li and Liu 43 ).
This study showed a very limited effect of L. paracasei F19 (9·4 × 1010 colony-forming units/dose) on the gut microbiota and metabolic markers, which were non-significant compared with the placebo group. The storage of L. paracasei F19 was optimised to ensure that the probiotic strain was alive at ingestion. The diary kept by the participants indicated that they were highly compliant to the intervention.
A combined effect of this probiotic strain on both metagenomic and metabolic markers in human subjects has not been explored previously. In mice, L. paracasei F19 has been shown to affect the immune system and expression of genes involved in energy homeostasis and insulin sensitivity( Reference Nerstedt, Nilsson and Ohlson 44 ), and to regulate body fat storage and lipoprotein metabolism( Reference Aronsson, Huang and Parini 45 ). Observational studies in human subjects have suggested that L. paracasei is associated with normal weight( Reference Million, Maraninchi and Henry 15 ) and negatively correlated with fasting blood glucose( Reference Stsepetova, Sepp and Kolk 16 ) despite that several other Lactobacillus species have been suggested to be associated with obesity( Reference Stsepetova, Sepp and Kolk 16 , Reference Armougom, Henry and Vialettes 46 , Reference Million, Angelakis and Paul 47 ) and T2D( Reference Karlsson, Tremaroli and Nookaew 7 ). It is possible that we would have observed an effect of the probiotic intervention if it had been administered as part of a fermentable milk product, as a fermentable food matrix could potentially enhance probiotic efficacy( Reference Sanders and Marco 48 ). A study among elderly has shown that daily intake of a fermented milk product with L. paracasei F19 (5·25 × 108 colony-forming units) over 4 weeks stimulates the growth of other Lactobacillus species in the gut( Reference Sullivan, Palmgren and Nord 49 ). However, no effect on other Lactobacillus species was observed following 6 weeks of intake of L. paracasei F19 in the present study, although it cannot be excluded that the abundance of Lactobacillus species not included in the gene catalogue did change( Reference Qin, Li and Raes 29 ).
The inclusion criteria in the present study were designed in order to recruit individuals at increased risk of metabolic diseases. Previous metagenomic studies have suggested that individuals with a metabolic risk profile are characterised by low gut microbiome richness( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ). The obese participants in these previous studies were comparable to the participants in the present study from a phenotype perspective; thus, it is surprising that only 8 % of the individuals in the present study had low gut microbiome richness as opposed to 23–40 % in the other cohorts( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ). The protocol for handling and analyses of microbiota samples were similar to the samples handling in the MetaHIT study( Reference Le Chatelier, Nielsen and Qin 5 ); however, it is possible that deviations in the prevalence of low microbiome richness between the cohorts can be explained by differences in habitual dietary habits. Yet, the negative correlations identified between bacterial gene count and metabolic risk markers support an association between gut microbiome richness and metabolic health. As does the lower microbiome richness detected at baseline in the participants allocated to L. paracasei F19, who were characterised by impaired insulin sensitivity, compared with the two other groups. This is interesting as the prediction model based on six bacterial species that has previously been demonstrated as a tool to identify individuals with low microbiome richness in two other obese cohorts( Reference Cotillard, Kennedy and Kong 4 , Reference Le Chatelier, Nielsen and Qin 5 ) also showed high specificity and sensitivity in this cohort.
In conclusion, the present study shows that intake of flaxseed mucilage improves insulin sensitivity and changes the gut microbiota in obese postmenopausal women, but suggests that the effect on insulin sensitivity is independent of the flaxseed mucilage-induced changes in abundance of bacterial species.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S0007114515001786
Acknowledgements
The authors thank the study participants; the kitchen staff, dieticians, laboratory technicians, good clinical practice coordinator and students involved in the project and the staff from the INRA/MetaGenoPolis: Florence Levenez and Joël Doré for sample preparation; Nathalie Galleron and Benoit Quinquis for microbiome sequencing; Jean-Michel Batto and Pierre Leonard for informatics. In addition to Sander Kersten, Division of Human Nutrition, Wageningen University, who kindly provided the protocol and guidance for the ANGPTL4 analyses. The study is part of The Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care (http://www.LuCAMP.org).
The present study was financially supported by The Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care (http://www.LuCAMP.org), and The Danish Dairy Board. Arla Foods amba delivered the probiotic product and provided additional funding. Metagenopolis is funded by the Investissement d'Avenir grant ANR-11-DPBS-0001.
The authors' contributions are as follows: L. K. B., T. H., O. P., A. A. and L. H. L. designed the research; L. K. B. and L. H. L. conducted the research; L. K. B., E. L. C., E. P., N. P., S. K., T. B., J. H., T. K. D., S. D. E. and L. H. L. contributed to the analysis and interpretation of the data; L. K. B. wrote the paper; L. K. B., E. L. C. and L. H. L. holds primary responsibility for the final content. All authors read and approved the final manuscript.
L. K. B., T. B., A. A. and L. H. L., as employed at the Department of Nutrition, Exercise and Sports, University of Copenhagen, received funding for research from the Arla Foods amba and the Danish Dairy Board. A. A. is a consultant or a member of the advisory boards for BioCare Copenhagen, DK; Basic Research, USA; Global Dairy Platform, USA; S-Biotek, DK. He holds rights to patents regarding properties of flaxseed registered in collaboration with the University of Copenhagen. J. H. is employed by Arla Strategic Innovation Centre, Stockholm, Sweden. E. L. C., E. P., N. P., S. K., T. K. D., T. H., O. P. and S. D. E. declared no conflicts of interest.








 Open access
Open access