


Flavanols, such as ( − )-epicatechin, catechin and their oligomers, represent a major class of flavonoids that are commonly present in most higher plants, and with high content in certain foods, such as grapes, tea and cocoa. Several epidemiological investigations and dietary interventions in human subjects using flavanol-containing foods indicate an inverse relationship between flavanol intake and the risk of CVD(Reference Duffy, Keaney and Holbrook1–Reference Deka and Vita5). A very wide range of biological actions of a flavanol-rich diet support these potential cardiovascular protective effects including the improvement of vasodilation(Reference Fisher, Hughes and Gerhard-Herman6–Reference Schroeter, Heiss and Balzer8), blood pressure(Reference Taubert, Berkels and Roesen9, Reference Matsuyama, Tanaka and Kamimaki10), insulin resistance(Reference Grassi, Lippi and Necozione11), the attenuation of platelet reactivity(Reference Holt, Lazarus and Sullards12), and the improvement of immune responses and antioxidant defence system(Reference Keen, Holt and Oteiza13). However, little is known about the molecular mechanisms of flavanol-mediated bioactivities in both humans and animals. The reasons for these shortcomings are, at least in part, based on the fact that food matrices contain a multitude of phytochemical constituents, of which an unknown number may exert physiological effects. The effect of high-flavanol cocoa was mimicked by oral intake of pure ( − )-epicatechin isolated from cocoa, and the maximum effect on endothelial function coincided with the peak of the plasma level of ( − )-epicatechin metabolites(Reference Schroeter, Heiss and Balzer8). ( − )-Epicatechin controls vascular tone in vitro inducing endothelium-dependent NO-mediated vasodilation(Reference Duarte, Perez Vizcaino and Utrilla14, Reference Huang, Zhang and Lau15) and improving endothelial dysfunction in diabetes(Reference Ihm, Lee and Kim16). ( − )-Epicatechin in vitro induced activation of endothelial NO synthase (eNOS) through two mechanisms: (i) eNOS phosphorylation by the participation of the phosphatidylinositol 3-kinase pathway and (ii) activation of the Ca2+/calmodulin-dependent kinase II pathway(Reference Ramirez-Sanchez, Maya and Ceballos17). Moreover, ( − )-epicatechin elevates NO in endothelial cells via inhibition of the main superoxide generating system NADPH oxidase(Reference Steffen, Gruber and Schewe18). Furthermore, a number of endothelial cell-protective actions of ( − )-epicatechin have been described, including protection against cytotoxicity, oxidative stress-related modifications of proteins and DNA, and proteasomal breakdown of eNOS(Reference Steffen, Gruber and Schewe18).
We hypothesised that chronic ( − )-epicatechin treatment might prevent the development of pathological changes associated with hypertension independently on its protective effects on NO. Therefore, we analysed the effects of ( − )-epicatechin, at doses equivalent to those achieved in the human diet, in a model of chronic inhibition of NO synthase (NOS) using the NOS inhibitor N G-nitro-l-arginine methyl ester (l-NAME). This model develops arterial hypertension and is associated with a vascular pro-oxidant, pro-atherogenic and proinflammatory phenotype(Reference Luvara, Pueyo and Philippe19, Reference Zatz and Baylis20).
Methods
Experimental protocols
The experimental protocol followed the European Union guidelines for animal care and protection. Male Wistar rats (315–400 g) were used in the study after an adaptation period of 2 weeks for vehicle administration and blood pressure measurements. Rats were randomly assigned to four different treatment groups for 4 weeks: (a) vehicle (control, 1 ml of 1 % methylcellulose once daily, n 8), (b) vehicle plus l-NAME (75 mg/100 ml in drinking-water, approximately 75 mg/kg per d, n 10), (c) ( − )-epicatechin (2 mg/kg inoculation by oral administration, mixed in 1 ml of 1 % methylcellulose once daily, n 10) plus l-NAME and (d) ( − )-epicatechin (10 mg/kg oral inoculation, mixed in 1 ml of 1 % methylcellulose once daily, n 10) plus l-NAME. The administration doses of ( − )-epicatechin in this study correspond to approximately 120–600 mg in humans (at a body weight of 60 kg), which are in the range than can be achieved in the human diet (e.g. flavanol content in dark chocolate used in intervention studies) and approximately two to ten times the average daily catechin intake in humans. The treatment with ( − )-epicatechin was stopped 2 d before the end of the study in order to analyse the long-term effects of ( − )-epicatechin without the involvement of the effects of acute administration; and on the last day, the animals were placed in metabolic cages to collect urine for 24 h.
Blood pressure measurements
Systolic blood pressure (SBP) was measured every week in conscious rats by tail-cuff plethysmography(Reference Duarte, Perez-Palencia and Vargas21). At least seven determinations were made in every session and the mean of the lowest three values within 5 mmHg was taken as the SBP.
Plasma and urinary determinations
Proteinuria was determined according to the Bradford method and the results were expressed as mg of protein excreted per 100 g of rat during 24 h. Plasma levels of malondialdehyde (MDA) were assessed following the method described by Esterbauer & Cheeseman(Reference Esterbauer and Cheeseman22). Total 8-iso-PGF2α (iso-PGF2α) and TNFα were measured by an enzyme-immunoassay kit (Cayman Chemical and Diaclone, Inc., Besancon, France, specific for rat TNFα, respectively).
Histological techniques
Formaldehyde-fixed, paraffin-embedded longitudinal kidney Wistar rat sections in the sagittal plane were stained with haematoxylin and eosin, and periodic acid-Schiff stain. The extent of vascular injury (stenosis, hyaline arteriopathy and myointimal proliferative hyperplasia) was assessed by examining profiles of arteries and arterioles in a single kidney section and counting the number of vessels affected. The presence of glomerular lesions (glomerulosclerosis and capsular fibrosis) was evaluated in at least 200 glomeruli. Tubular atrophy and tubular casts were also evaluated. The morphological study was done in blinded fashion on 4-μm sections with light microscopy, using the most appropriate stain for each lesion. The values were expressed as the percentage of rats with lesions in each group, and the severity of lesions was calculated semiquantitatively using a 0-to-3 scale (0, absence; 1, mild ( < 10 % of vessel, tubules or glomeruli involved); 2, moderate (10–25 %); 3, severe (>25 %)).
Vascular contractility in vitro
Descending thoracic aortic rings (3 mm) and the fourth branch of the mesenteric artery (1·7–2 mm) were dissected from animals and were mounted in organ chambers and in a wire myograph (model 610M; Danish Myo Technology, Aarhus, Denmark), respectively, filled with Krebs solution as previously described(Reference Duarte, Ocete and Perez-Vizcaino23). In endothelium-denuded aorta, the concentration–relaxation response curves to nitroprusside (10− 9–10− 5 m) were performed in rings pre-contracted by 10− 6 m-phenylephrine. The relaxant responses to acetylcholine were also studied in both intact-small mesenteric artery and -aorta pre-contracted by phenylephrine (5 μm in mesenteric artery and 1 or 0·1 μm in control or l-NAME-treated aortic rings, respectively, to obtain a similar level of pre-contraction). Contractions evoked by acetylcholine were tested in aortic rings with endothelium incubated for 30 min with l-NAME (10− 4 m); responses were expressed as a percentage of a previous response to 80 mm-KCl.
In situ detection of vascular superoxide anion levels
Unfixed thoracic aortic rings were cryopreserved (PBS 0·1 mol/l, plus 30 % sucrose for 1–2 h), included in optimum cutting temperature, frozen ( − 80°C), and 10 μm cross-sections were obtained in a cryostat. Sections were incubated for 30 min in HEPES-buffered solution containing dihydroethidium (10− 5 m), counterstained with the nuclear stain 4′-6-diamidino-2-phenylindole and photographed on a fluorescence microscope. Superoxide anion (![]() ) level was estimated from the ratio of ethidium:4′-6-diamidino-2-phenylindole fluorescence(Reference Vera, Sanchez and Galisteo24). In preliminary experiments, dihydroethidium fluorescence was almost abolished by the
) level was estimated from the ratio of ethidium:4′-6-diamidino-2-phenylindole fluorescence(Reference Vera, Sanchez and Galisteo24). In preliminary experiments, dihydroethidium fluorescence was almost abolished by the ![]() scavenger tiron, indicating the specificity of this reaction.
scavenger tiron, indicating the specificity of this reaction.
NADPH oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine NADPH oxidase activity in intact aortic rings, as previously described(Reference Sanchez, Lodi and Vera25). Aortic production of ![]() was stimulated by the addition of NADPH (100 μm). Rings were then placed with or without NADPH, and lucigenin was injected automatically at a final concentration of 5 μm to avoid known artifacts when used at higher concentrations in a scintillation counter (Lumat LB 9507; Berthold, Germany). Vessels were then dried, and dry weight was determined. NADPH oxidase activity is expressed as relative luminescence units/min per mg dry aortic tissue.
was stimulated by the addition of NADPH (100 μm). Rings were then placed with or without NADPH, and lucigenin was injected automatically at a final concentration of 5 μm to avoid known artifacts when used at higher concentrations in a scintillation counter (Lumat LB 9507; Berthold, Germany). Vessels were then dried, and dry weight was determined. NADPH oxidase activity is expressed as relative luminescence units/min per mg dry aortic tissue.
Western blotting analysis
Aortic homogenates were run on SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membranes, incubated with primary monoclonal mouse anti-eNOS (Cell Signaling Technology, Danvers, MA, USA), anti-phospho-eNOS (Ser-1177) (Cell Signaling Technology), rabbit polyclonal anti-cyclo-oxygenase 2 (COX-2; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-phospho-Akt-Ser-473, rabbit anti-Akt, rabbit anti-extracellular-signal-regulated kinase 1/2 (ERK1/2; Cell Signaling Technology) or mouse anti-phospho-ERK1/2-Thr183 and Tyr185 (Sigma-Aldrich, Alcobendas, Spain), polyclonal goat anti-p22phox, or polyclonal rabbit anti-p47phox (Santa Cruz Biotechnology) antibodies overnight and with the correspondent secondary peroxidase conjugated antibodies. Antibody binding was detected by an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Amersham, UK) and densitometric analysis was performed using Scion Image-Release Beta 4.02 software (Scion Corporation, Frederick, MD, USA). Samples were re-probed for expression of smooth muscle α-actin or ERK1/2. Protein abundance:α-actin ratio or phospho-Akt/Akt, phospho-ERK1/2/ERK1/2 and phospho-eNOS/eNOS were calculated and data are expressed as a percentage of the values in control aorta from the same gel.
RT-PCR analysis
For RT-PCR analysis, total RNA was extracted and converted to complementary DNA by standard methods. PCR was performed with a Techne Techgene thermocycler (Techne, Cambridge, UK). Initial denaturation was done at 95°C for 3 min and followed by thirty-two to thirty-five (thirty-two for p22phox, eNOS, intercellular adhesion molecule-1 (ICAM-1), TNFα and IL-1β, and thirty-five for p47phox) cycles of amplification. Each cycle consisted of 1 min of denaturation at 94°C, 45 s of annealing at 55°C for p22phox, ICAM-1 and IL-1β, 57°C for TNFα and 58°C for eNOS and 57°C for p47phox, and 1 min for enzymatic primer extension at 72°C. After the final cycle, the temperature was held at 72°C for 10 min to allow reannealing of amplified products. RT-PCR products were then size-fractionated through a 1 % agarose gel, and the bands were visualised with ethidium bromide and quantified by densitometric analysis performed on the scanned images using Scion Image-Release Beta 4.02 software (Scion Corporation, Frederick, MD, USA; http://www.scioncorp.com). The sequences for primers were as follows: p47phox (100 bp) sense, 5′-ATGACAGCCAGGTGAAGAAGC-3′ and antisense, 5′-CGATAGGTCTGAAGGCTGATGG-3′; p22phox (220 bp) sense 5′-GCGGTGTGGACAGAAGTACC-3′ and antisense, 5′-CTTGGGTTTAGGCTCAATGG-3′; eNOS (161 bp) sense 5′-ATGGATGAGCCAACTCAAGG-3′ and antisense, 5′-TGTCGTGTAATCGGTCTTGC-3′; ICAM-1 (386 bp) sense 5′-AGGTATCCATCCATCCCACA-3′ and antisense, 5′-AGTGTCTCATTGCCACGGAG-3′; TNFα (468 bp) sense 5′-ATGTGGAACTGGCAGAGGAG-3′ and antisense, 5′- GGCCATGGAACTGATGAGAG-3′; IL-1β (497 bp) sense 5′- AGGCAGTGTCACTCATTGTG-3′ and antisense, 5′-GGAGAGCTTTCAGCTCACAT-3′. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control for the co-amplification. The signals were expressed relative to the intensity of glyceraldehyde 3-phosphate dehydrogenase in each sample.
Drugs
All drugs and chemicals were obtained from Sigma (Alcobendas, Spain), and dissolved in distilled deionised water, except for ( − )-epicatechin, which was mixed with 1 % methylcellulose.
Statistical analysis
Results are expressed as means with their standard errors. Statistically significant differences were calculated by one-way ANOVA analysis followed by Bonferroni's post hoc test. P < 0·05 was considered statistically significant. Renal lesion severity was analysed by the Mann–Whitney U test.
Results
Effects in blood pressure and left ventricular and kidney weight indices
Rats receiving chronic l-NAME treatment showed a progressive increase in SBP and decrease in heart rate (Fig. 1), which were already significant after the first week. Concomitant treatment with ( − )-epicatechin (2 and 10 mg/kg) did not prevent the changes in either SBP or heart rate. At the end of the study period, l-NAME significantly increased the heart (13 %) and left ventricular (22 %) weight indices as compared to the control group. In rats receiving l-NAME plus ( − )-epicatechin at 10 mg/kg, these parameters were significantly reduced as compared to the l-NAME group (Table 1).

Fig. 1 Effects in (a) systolic blood pressure (SBP) and (b) heart rate (HR) as measured by tail-cuff plethysmography in control (□), N G-nitro-l-arginine methyl ester (l-NAME, ■), l-NAME+2 mg/kg epicatechin (Epi 2, ○) and l-NAME+10 mg/kg epicatechin (Epi 10, ●) groups. Values are means, with their standard errors represented by vertical bars. Mean values were significantly different between l-NAME and control group: *P < 0·05, **P < 0·01. bpm, beats per minute.
Table 1 Body weight (BW) and cardiac and renal indices
(Mean values with their standard errors)
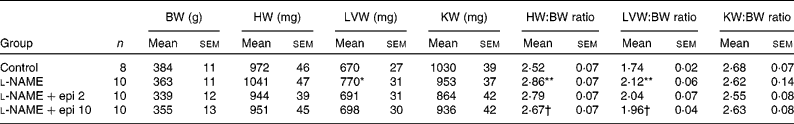
HW, heart weight; LVW, left ventricular weight; KW, kidney weight; l-NAME, N G-nitro-l-arginine methyl ester; epi, epicatechin.
Mean values were significantly different from those of the control group: *P < 0·01, **P < 0·01.
† Mean values were significantly different from those of the l-NAME group (P < 0·05).
Effects in renal histology and proteinuria
l-NAME induced moderate/severe renal injury which affected 40 % of the animals. Hyaline and proliferative (myointimal proliferative hyperplasia) arteriopathy were the main and most intense lesions associated with thickening of the vascular wall and decrease of lumen (Fig. 2(a) and Table S1, supplementary material for this article can be found at http://www.journals.cambridge.org/bjn). The intensity, number and size of the vessel affected with hyaline arteriopathy and myointimal proliferative hyperplasia were significantly decreased by treatment with ( − )-epicatechin 10 mg/kg. Glomerular and tubulointerstitial lesions were not present, except in l-NAME and l-NAME-plus epicatechin 2 groups which present only weak and scattered tubular cast and tubular atrophy. Proteinuria was markedly increased in the l-NAME group and this effect was reduced only by 10 mg/kg ( − )-epicatechin (Fig. 2(b)).

Fig. 2 Effects of epicatechin (Epi) in renal injury. (a) Renal parenchyma in N G-nitro-l-arginine methyl ester (l-NAME) hypertension model. (i) Absence of vascular, glomerular or tubulointerstitical lesions in control (C) group; (ii) vessels with hyaline arteriopathy and myointimal proliferative hyperplasia (arrows) in l-NAME group; (iii) moderate/severe hyaline arteriopathy in afferent arteriole of glomerulus (*) and interlobular arteria with lumen reduction (arrow) in l-NAME+Epi 2 group; (iv) circumferential hyalin arteriopathy without lumen reduction in l-NAME+Epi 10 group (arrow). (b) Proteinuria in all experimental groups. Values are means, with their standard errors represented by vertical bars. * Mean values were significantly different between the l-NAME and C groups (P < 0·05). † Mean values were significantly different between the l-NAME-Epi and l-NAME groups (P < 0·05).
Effects in systemic reactive oxygen species
Plasma MDA level, a marker of lipid peroxidation induced by reactive oxygen species (ROS), in l-NAME-treated animals was increased (89 %) as compared to the control group. In l-NAME plus ( − )-epicatechin-treated rats, MDA concentration was reduced only in rats treated with 10 mg/kg ( − )-epicatechin (Fig. 3(a)). The 24 h urinary iso-PGF2α excretion, a more specific marker of oxidative stress, was also increased in the L-NAME group. In both groups of ( − )-epicatechin-treated l-NAME rats, iso-PGF2α excretion showed similar values to those of control rats (Fig. 3(b)).

Fig. 3 Effects of epicatechin (Epi) in systemic oxidative markers. (a) Plasma malondialdehyde (MDA) content and (b) urinary excretion of 8-iso-PGF2α in control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Values are means, with their standard errors represented by vertical bars. Mean values were significantly different between the l-NAME and C groups: *P < 0·05, **P < 0·01. Mean values were significantly different between the l-NAME-Epi and l-NAME groups: †P < 0·05; ††P < 0·01. BW, body weight.
Effects in vascular NO pathway
Aortae from l-NAME-treated rats showed strongly reduced endothelium-dependent vasodilator responses to acetylcholine (Fig. 4(a)). The aortae from l-NAME plus ( − )-epicatechin at 10 mg/kg treated animals showed a small increase in the vasodilation induced by acetylcholine as compared to animals from the l-NAME group. In aortic rings, no differences were observed among groups in the endothelium-independent vasodilator responses to the NO donor sodium nitroprusside (Fig. 4(b)). In intact-small mesenteric arteries, acetylcholine elicited concentration-dependent relaxations that were inhibited by chronic l-NAME treatment (Fig. 4(c)). ( − )-Epicatechin 2 mg/kg did not prevent this effect of l-NAME and the dose of 10 mg/kg marginally but significantly increased the relaxant response induced by the highest concentration of acetylcholine.

Fig. 4 Effects of epicatechin (Epi) on endothelial function. Vascular relaxant responses induced by (a) acetylcholine (ACh) and (b) sodium nitroprusside (SNP) in aortae pre-contracted by 1 μm phenylephrine (Phe), and by ACh in small mesenteric arteries contracted by 5 μm-Phe (c) in aortae from control (□), N G-nitro-l-arginine methyl ester (l-NAME, ■), l-NAME+2 mg/kg Epi (Epi 2, ○) and l-NAME+10 mg/kg Epi (Epi 10, ●) groups. Values are means, with their standard errors represented by vertical bars. Mean values were significantly different between the l-NAME and control groups: *P < 0·05, **P < 0·01. † Mean values were significantly different between the l-NAME-Epi and l-NAME groups (P < 0·05).
eNOS mRNA in the aorta from the l-NAME group was increased as compared to the control group and both groups of rats treated with l-NAME plus ( − )-epicatechin showed reduced expression of eNOS as compared to the l-NAME group alone (Fig. 5(b)). We also observed that ( − )-epicatechin treatment significantly increased eNOS phosphorylation at Ser-1177, as compared to the control and the l-NAME groups (Fig. 5(c)).

Fig. 5 Effects of epicatechin (Epi) on endothelial nitric oxide synthase (eNOS). Gene expression of eNOS by (a) RT-PCR, (b) Western blot and (c) Ser-1177-phospho-eNOS (p-eNOS) in control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Panels show representative bands and histograms represent densitometric values normalised to the corresponding RT-PCR products of (a) glyceraldehyde 3-phosphate dehydrogenase (GADPH) or normalised to the corresponding (b) α-actin or (c) eNOS. Values are means, with their standard errors represented by vertical bars (n 3–5). **Mean values were significantly different between the l-NAME and C groups (P < 0·01). Mean values were significantly different between the l-NAME-Epi and l-NAME groups: †P < 0·05; ††P < 0·01.
Effects on vascular cyclo-oxygenase pathway
Aortae from l-NAME-treated rats showed strongly increased endothelium-dependent vasoconstrictor responses to acetylcholine (Fig. 6(a)), which are related to COX-derived metabolites. This effect was accompanied by an increased COX-2 mRNA (Fig. 6(b)) and protein expression (Fig. 6(c)). The aortae from l-NAME plus ( − )-epicatechin at 10 mg/kg treated animals showed reduced vasoconstrictor responses to acetylcholine and COX-2 expression, as compared to the l-NAME group.

Fig. 6 Effects of epicatechin (Epi) on the cyclo-oxygenase (COX) pathway. (a) Endothelium-dependent contractions induced by acetylcholine (ACh) in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. ACh-induced contractions were induced in arteries treated with l-NAME (10− 4 m) in the organ bath and expressed as a percentage of the response to 80 mm-KCl. Expression of (b) COX-2 at the level of mRNA by RT-PCR and (c) protein by Western blot in all experimental groups. Panels show representative bands and histograms represent densitometric values normalised to the corresponding RT-PCR products of (b) glyceraldehyde 3-phosphate dehydrogenase (GADPH) or normalised to the corresponding (c) α-actin. Values are means, with their standard errors represented by vertical bars (n 3–5). **Mean values were significantly different between the l-NAME and C groups (P < 0·01). † Mean values were significantly different between the l-NAME-Epi and l-NAME groups (P < 0·05).
Effects in vascular superoxide anion production and NADPH oxidase pathway
To characterise and localise vascular ROS production, sections of aorta were incubated with dihydroethidium which is oxidised by ![]() to yield the red fluorescent DNA stain ethidium. Fluorescence was almost suppressed by the
to yield the red fluorescent DNA stain ethidium. Fluorescence was almost suppressed by the ![]() scavenger tiron (10 mM, data not shown). Rings from l-NAME rats showed marked increased staining in adventitial, medial and endothelial cells (Fig. 7(a)). Red fluorescence, normalised to the blue fluorescence of the nuclear stain 4′-6-diamidino-2-phenylindole, was reduced after 10 mg/kg ( − )-epicatechin treatment (Fig. 7(b)).
scavenger tiron (10 mM, data not shown). Rings from l-NAME rats showed marked increased staining in adventitial, medial and endothelial cells (Fig. 7(a)). Red fluorescence, normalised to the blue fluorescence of the nuclear stain 4′-6-diamidino-2-phenylindole, was reduced after 10 mg/kg ( − )-epicatechin treatment (Fig. 7(b)).

Fig. 7 Effects of epicatechin (Epi) in aortic superoxide anion (![]() ) levels. (a) Left pictures show arteries incubated in the presence of dihydroethidium which produces a red fluorescence when oxidised to ethidium by
) levels. (a) Left pictures show arteries incubated in the presence of dihydroethidium which produces a red fluorescence when oxidised to ethidium by ![]() . Right pictures show blue fluorescence of the nuclear stain 4′-6-diamidino-2-phenylindole (DAPI) (400 × magnification). (b) Averaged values, mean with their standard errors (n 5–7 rings from different rats) of the red ethidium fluorescence normalised to the blue DAPI fluorescence in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Values are means, with their standard errors represented by vertical bars. **Mean values were significantly different between the l-NAME and C groups (P < 0·01). Mean values were significantly different between the l-NAME-Epi and l-NAME groups: †P < 0·05; ††P < 0·01.
. Right pictures show blue fluorescence of the nuclear stain 4′-6-diamidino-2-phenylindole (DAPI) (400 × magnification). (b) Averaged values, mean with their standard errors (n 5–7 rings from different rats) of the red ethidium fluorescence normalised to the blue DAPI fluorescence in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Values are means, with their standard errors represented by vertical bars. **Mean values were significantly different between the l-NAME and C groups (P < 0·01). Mean values were significantly different between the l-NAME-Epi and l-NAME groups: †P < 0·05; ††P < 0·01.
NADPH increases lucigenin luminescence in normal aortic rings, which was almost abolished by the flavoprotein inhibitor diphenyliodonium (10 μm, not shown), showing that external NADPH increased NADPH oxidase activity in vascular tissue. NADPH oxidase activity was increased in aortic rings from l-NAME rats as compared to control rats. Chronic treatments with ( − )-epicatechin, at both doses, abolished this increased NADPH oxidase activity in l-NAME-treated rats (Fig. 8(a)). Significant p22phox mRNA (Fig. 8(b)) and protein (Fig. 8(c)) up-regulation, without changes in p47phox (Fig. 8(d) and (e)), was observed in aortic tissue from l-NAME rats. ( − )-Epicatechin treatment, at both doses, inhibited the p22phox gene overexpression in l-NAME-treated animals.

Fig. 8 Effects of epicatechin (Epi) in the NADPH oxidase pathway. (a) NADPH oxidase activity measured by lucigenin-enhanced chemiluminescence, and expression of NADPH oxidase subunits p22phox and p47phox at the level of (b and d) mRNA by RT-PCR and (c and e) protein by Western blot in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Panels show representative bands and histograms represent densitometric values normalised to the corresponding RT-PCR products of (b and d) glyceraldehyde 3-phosphate dehydrogenase (GADPH) or normalised to the corresponding (c and e) α-actin. Values are means, with their standard errors represented by vertical bars (n 3–5). Mean values were significantly different between the l-NAME and C groups: *P < 0·05, **P < 0·01. † Mean values were significantly different between the l-NAME-Epi and l-NAME groups (P < 0·05). RLU, relative luminescence units.
Effects on inflammatory status
The mRNA expression of ICAM-1 (Fig. 9(a)) and proinflammatory cytokines IL-1β (Fig. 9(b)), or TNFα (Fig. 9(c)) in aortic homogenates was higher in aortae from l-NAME groups as compared to control rats. Plasma TNFα levels (Fig. 9(d)) were also increased in l-NAME-treated rats. ( − )-Epicatechin treatment, at both doses, significantly down-regulated these genes, and reduced plasma levels of TNFα.

Fig. 9 Effects of epicatechin (Epi) in proinflammatory genes. Panels show representative bands and histograms represent densitometric values normalised to the corresponding RT-PCR products of (a) glyceraldehyde 3-phosphate dehydrogenase (GADPH) for intercellular adhesion molecule-1 (ICAM-1), (b) IL-1 Iβ and (c) TNFα (n 3–5) in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. (d) Plasma TNFα levels in all experimental groups. Values are means, with their standard errors represented by vertical bars. ** Mean values were significantly different between the l-NAME and C groups (P < 0·01). Mean values were significantly different between the l-NAME-Epi and l-NAME groups: †P < 0·05, ††P < 0·01.
Effects on vascular extracellular-signal-regulated kinase 1/2 and Akt pathways
The expression of phospho-protein kinase B (Akt) (Fig. 10(a)) and phospho-ERK1/2 (Fig. 10(b)) proteins was increased in aorta by l-NAME. Chronic ( − )-epicatechin treatment increased Akt phosphorylation and reduced ERK1/2 phosphorylation in rats with chronic NO-deficient hypertension.

Fig. 10 Effects of epicatechin (Epi) in protein kinase B (Akt) and extracellular-signal-regulated kinase (ERK) pathways. (a and b) Representative bands and histograms represent densitometric values of phospho-Akt and phospho-ERK1/2 (p-ERK1/2) relative to total Akt and ERK1/2 protein levels (n 3–5) in aortae from control (C), N G-nitro-l-arginine methyl ester (l-NAME), l-NAME+2 mg/kg Epi (Epi 2) and l-NAME+10 mg/kg Epi (Epi 10) groups. Values are means, with their standard errors represented by vertical bars. * Mean values were significantly different between the l-NAME and C groups (P < 0·05). † Mean values were significantly different between the l-NAME-Epi and l-NAME groups (P < 0·05).
Discussion
Administration of l-NAME in drinking-water induces a progressive increase in arterial blood pressure which is attributed to a reduced synthesis of the vasodilator NO and has been widely used as a model of chronic hypertension(Reference Zatz and Baylis20). The present study shows for the first time that a single oral daily dose of ( − )-epicatechin (2 or 10 mg/kg) partially or fully prevented most of the effects induced by l-NAME (Fig. 11) such as (a) increases in the left ventricular hypertrophy, (b) proteinuria, (c) renal histological lesions, (d) increased plasma MDA concentrations and urinary iso-PGF2α excretion, (e) increased endothelium-dependent contraction and COX-2 overexpression, (f) increased vascular production of ![]() and NADPH oxidase activity and (g) increased vascular inflammatory status. In most cases, these effects were dose-dependent. However, it did not inhibit the development of hypertension and had only minor effects on the impaired endothelium- and NO-dependent relaxation. Interestingly, changes in several end-points were not dependent on the presence of ( − )-epicatechin in blood, since they were obtained 48 h after the deprivation of the flavanol, indicating that it alters the course of the disease via permanent structural changes and/or alteration of gene expression.
and NADPH oxidase activity and (g) increased vascular inflammatory status. In most cases, these effects were dose-dependent. However, it did not inhibit the development of hypertension and had only minor effects on the impaired endothelium- and NO-dependent relaxation. Interestingly, changes in several end-points were not dependent on the presence of ( − )-epicatechin in blood, since they were obtained 48 h after the deprivation of the flavanol, indicating that it alters the course of the disease via permanent structural changes and/or alteration of gene expression.

Fig. 11 Schematic diagram representing the mechanism of action of epicatechin (Epi) on the inflammatory and vascular dysfunction pathways in nitric oxide (NO)-deficient hypertensive rats. l-NAME, N G-nitro-l-arginine methyl ester; eNOS, endothelial NO synthase; p-eNOS, phospho-eNOS; COX-2, cyclo-oxygenase 2; ![]() , superoxide anion; ERK1/2, extracellular-signal-regulated kinase 1/2.
, superoxide anion; ERK1/2, extracellular-signal-regulated kinase 1/2.
( − )-Epicatechin, at concentrations >30 μm, exhibits vasodilator effects in vitro, which are partially endothelium- and NO-dependent(Reference Duarte, Perez Vizcaino and Utrilla14, Reference Huang, Zhang and Lau15). ( − )-Epicatechin activates eNOS in human coronary artery endothelial cells by (i) Ser-633 and Ser-1170 phosphorylation and Thr-495 dephosphorylation, and (ii) via Ca2+/calmodulin-dependent kinase II pathways, leading to increased NO production(Reference Ramirez-Sanchez, Maya and Ceballos17). Moreover, ( − )-epicatechin and its two in situ O-methylated metabolites elevate NO in endothelial cells via inhibition of NADPH oxidase(Reference Steffen, Gruber and Schewe18). However, it is predictable that this acute effect was absent in vivo in animals treated with l-NAME because: (1) the concentrations of ( − )-epicatechin in plasma after 2 or 10 mg/kg ( − )-epicatechin(Reference Piskula and Terao26) are below its active range of concentrations as vasodilators and (2) its relaxant response would be small under conditions of inhibited eNOS. Therefore, the lack of antihypertensive effect of ( − )-epicatechin in this model of hypertension may be related, at least in part, to the absence of acute vasodilator effect.
Renal and cardiac injury
Renal injury has been consistently reported after chronic inhibition of NO synthesis(Reference Zatz and Baylis20). In our study, the l-NAME group presented moderate/severe kidney injury, especially in the vasculature, and with low tubular casts and mild tubular atrophy. The main and most intense vascular lesion in l-NAME rats was hyaline arteriopathy and thickening of vascular wall (proliferative arteriopathy) with moderate decrease of lumen, which was observed almost always in medium-sized vessels. These histological findings were associated with proteinuria, indicating functional impairment of the glomerular wall barrier. ( − )-Epicatechin, at the higher dose used, partially prevented renal parenchyma and vascular lesions and proteinuria, indicating that ( − )-epicatechin protects, at least partially, from l-NAME-induced renal injury, despite the lack of antihypertensive effect. A modest left ventricular hypertrophy has also been found in this model of hypertension(Reference Bartunek, Weinberg and Tajima27). In our study, the heart and left ventricular weight indices were significantly increased in l-NAME-treated rats and these effects were significantly prevented by 10 mg/kg ( − )-epicatechin. These protective effects would be at least partly due to a reduction in ROS and proinflammatory cytokines induced by ( − )-epicatechin, which are potent stimulus for cardiac growth and renal injury(Reference Li, Gall and Grieve28, Reference Vaziri29).
Endothelial dysfunction, NO and cyclo-oxygenase-derived vasoconstrictors
Administration of l-NAME is associated with endothelial dysfunction(Reference Zatz and Baylis20). As expected, l-NAME-treated rats showed reduced endothelium-dependent vasodilator responses induced by acetylcholine in both small arteries and in aorta, with similar endothelium-independent relaxant response to the NO donor nitroprusside. ( − )-Epicatechin 10 mg/kg only weakly prevented this effect in both vascular beds, without affecting NO sensitivity, since it did not modify the vasodilation induced by nitroprusside.
l-NAME inhibits the constitutive Ca2+-dependent NOS isoforms (eNOS and nNOS) and the Ca2+-independent inducible NOS isoform. However, these inhibitory effects on NOS activity were associated with increased eNOS expression in aortic tissues, which may be viewed as a compensatory mechanism to maintain the production of bioactive NO in the face of increased oxidant stress(Reference Zhen, Lu and Wang30). ( − )-Epicatechin reduced eNOS gene and protein overexpression, possibly as a result of the reduced vascular superoxide levels in the aortae. Interestingly, we also found an increased eNOS phosphorylation of Ser1177, associated with an increased vascular Akt phosphorylation in both groups of rats treated with ( − )-epicatechin. These in vivo results are consistent with previous in vitro observations showing ( − )-epicatechin-induced eNOS activation via PI3K/Akt-mediated phosphorylation in human endothelial cells(Reference Ramirez-Sanchez, Maya and Ceballos17).
In addition, increased endothelium-dependent vasoconstriction induced by acetylcholine in the presence of l-NAME in the organ chamber was also observed in aorta from l-NAME-treated rats. These contractions have been previously attributed to increased endothelial release of COX-derived vasoconstrictor prostanoids (such as PGH2 or thromboxane A2)(Reference Auch-Schwelk, Katusic and Vanhoutte31, Reference Duarte, Jimenez and O'Valle32). Likewise, COX-2 overexpression was found in aorta from l-NAME-treated rats. ( − )-Epicatechin at 10 mg/kg reduced acetylcholine-induced vasoconstriction and prevented the increase in COX-2, suggesting that ( − )-epicatechin inhibits the release of COX-derived metabolites by down-regulating COX-2. Thus, although chronic ( − )-epicatechin prevented the secondary changes in endothelial function (due to increased release of endothelial vasoconstrictors), it was unable to restore the primary endothelial defect (i.e. deficient NO production).
Systemic and vascular reactive oxygen species and inflammatory markers
ROS have been suggested to contribute to the genesis of atherosclerosis, diabetes, IHD, heart failure and hypertension. The l-NAME model of hypertension has been also associated with increased systemic oxidative stress(Reference Duarte, Jimenez and O'Valle32). In fact, plasma MDA values which reflect a general index of the oxidative status and lipid peroxidation and the 24 h urinary levels of isoprostane F2α, a PG-like compound produced by the reaction of arachidonic acid and superoxide(Reference Belik, Gonzalez-Luis and Perez-Vizcaino33), were increased in the present study. ( − )-Epicatechin in vitro is known to possess antioxidant properties(Reference van Acker, van den Berg and Tromp34) and reduces the MDA content in erythrocytes from hypertensive patients(Reference Kumar, Kant and Maurya35). In agreement with these data, in our experiment, long-term ( − )-epicatechin treatment reduced plasma MDA levels and the urinary levels of isoprostanes.
In agreement with other models of hypertension(Reference Sanchez, Galisteo and Vera36), hypertension induced by chronic blockade of NO production was associated with an overproduction of ROS also within the arterial wall(Reference Gonzalez, Fontaine and Pueyo37). In our in situ detection of ![]() production study, we found that rings from the l-NAME group showed marked staining as compared to control rats, and it was distributed in all layers of the aortic wall. This overproduction may be due, at least in part, to an increase in NADPH oxidase activity, as suggested by both NADPH-stimulated lucigenin-enhanced chemiluminescence and an increase in p22phox expression (present results and Gonzalez et al. (Reference Gonzalez, Fontaine and Pueyo37)). ( − )-Epicatechin, at both doses, strongly reduced ethidium fluorescence, NADPH oxidase activity and p22phox up-regulation in aortic rings from l-NAME-treated rats. Similar results in the vascular wall were also described using a higher dose (30 mg/kg) of its stereoisomer catechin in prediabetic Otsuka Long Evans Tokushima Fatty rats(Reference Ihm, Lee and Kim16). In our study, chronic NO inhibition also induced increased ERK1/2 phosphorylation, an effect which was prevented by ( − )-epicatechin and may be involved in its inhibitory effects on NADPH oxidase activity.
production study, we found that rings from the l-NAME group showed marked staining as compared to control rats, and it was distributed in all layers of the aortic wall. This overproduction may be due, at least in part, to an increase in NADPH oxidase activity, as suggested by both NADPH-stimulated lucigenin-enhanced chemiluminescence and an increase in p22phox expression (present results and Gonzalez et al. (Reference Gonzalez, Fontaine and Pueyo37)). ( − )-Epicatechin, at both doses, strongly reduced ethidium fluorescence, NADPH oxidase activity and p22phox up-regulation in aortic rings from l-NAME-treated rats. Similar results in the vascular wall were also described using a higher dose (30 mg/kg) of its stereoisomer catechin in prediabetic Otsuka Long Evans Tokushima Fatty rats(Reference Ihm, Lee and Kim16). In our study, chronic NO inhibition also induced increased ERK1/2 phosphorylation, an effect which was prevented by ( − )-epicatechin and may be involved in its inhibitory effects on NADPH oxidase activity.
Monocyte recruitment is one of the early steps in hypertension-induced arteriosclerosis and perivascular fibrosis(Reference Nicoletti and Michel38). Chronic NO suppression induced an increase in ex vivo monocyte endoluminal adhesion and in in vivo perivascular macrophage accumulation, in concert with increases in oxidative stress and inflammatory cytokines in the arterial wall(Reference Luvara, Pueyo and Philippe19). IL-1β, ICAM-1 and TNFα mRNA levels were increased in the vascular wall of the l-NAME-treated rats, suggesting that proinflammatory signals come from the arterial wall. The expression of adhesion molecules and proinflammatory cytokines are mainly the products of inducible genes that are usually controlled, at least in part, by the redox-sensitive NF-κB pathway(Reference Brand, Page and Walli39). The increase in oxidative stress in the vascular wall of l-NAME-treated rats probably activates the NF-κB system, which, in turn, induces the expression of proinflammatory cytokines(Reference Hernandez-Presa, Bustos and Ortego40). Our results further support this hypothesis, since ( − )-epicatechin, which reduced aortic superoxide levels also inhibited the vascular expression of these proinflammatory and proatherogenic markers.
In the present model in which NO bioactivity is impaired, no change in blood pressure and only marginal effects on endothelial function were found with ( − )-epicatechin. This contrasts with human intervention studies with tea(Reference Duffy, Keaney and Holbrook1) and with the effects of epicatechin in other animal models of hypertension (M Gomez-Guzman, R Jimenez and J Duarte, unpublished results) indicating that NO plays an essential role in the effects of ( − )-epicatechin on blood pressure and endothelial function. Our results are consistent with favourable effects of tea and other flavanol-containing food on cardiovascular risk factors(Reference Deka and Vita5). However, caution should be taken with the potential use of green tea extracts as supplements, rich in ( − )-epicatechin, because of reports of potential liver damage at high doses.
In conclusion, the present study demonstrates that chronic ( − )-epicatechin treatment, at doses equivalent to those that can be achieved in the human diet, prevented cardiac hypertrophy, renal parenchyma and vascular lesions and proteinuria, and blunted the prostanoid-mediated enhanced endothelium-dependent vasoconstrictor responses in the model of chronic inhibition of NO synthesis with l-NAME. Furthermore, ( − )-epicatechin also reduced the vascular oxidative stress and proinflammatory status, early events involved in atherosclerosis development.
Acknowledgements
The present study was supported by Grants from Comisión Interministerial de Ciencia y Tecnología (AGL2007-66108/ALI, SAF2007-62731, SAF2008-03948, SAF2010-22066-C02-01), from Junta de Andalucía, Proyecto de Excelencia (P06-CTS-01555) and by the Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III (Red HERACLES RD06/0009), Spain. M. J. Z., P. G., M. R., R. L.-S., A. M. Q., M. G.-G. and E. B. are holders of studentships from Spanish Ministry of Science and Education. The authors declare no conflicts of interest. J. D. and R. J. designed the experiments, J. D. and F. P.-V. wrote the manuscript, and all others performed and analysed the experiments and contributed to the discussion.












