A 71-year-old right-handed woman developed global aphasia and right hemiparesis at wake-up, last seen normal 14 hours ago. Her medical history included high blood pressure, dyslipidemia, diabetes, and coronary artery bypass graft one month prior to her presentation. NIHSS score was 18. Cerebral CT-scan (Figure 1A) showed no signs of acute infarction. CT-angiography revealed a terminal intracranial carotid occlusion (Figure 1B) with no ipsilateral carotid stenosis. She was transferred to the nearest comprehensive stroke center for endovascular therapy (EVT). Complete recanalization (TICI 3) was obtained 5 hours after presentation using a stent-retriever device with thrombo-aspiration. She improved in the following hours (NIHSS 2). There were no signs of infarction on follow-up brain CT-scan at 24 hours (Figure 1C). Transthoracic heart echocardiogram was normal. She was discharged home on day 8 with no residual clinical deficit. Post-heart surgery paroxysmal atrial fibrillation was suspected. Prolonged cardiac monitoring was ordered on an outpatient basis.

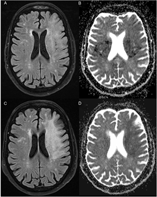
Figure 1: (A) Cerebral CT-scan at initial presentation shows no signs of acute infarction (ASPECTS 10). (B) Cerebral angio-CT reveals an occlusion of the termination of the left intracranial carotid artery (white arrow). (C) Cerebral CT-scan 24 hours after EVT remains unchanged, with no evidence of infarction.
She was re-admitted 10 days later with recurrence of aphasia and right hemiparesis, following a progressive non-fluctuating course over the past 3 days. There was no evidence of seizure activity or intercurrent illness. Her blood pressure was stable and normal. Brain MRI identified T2/FLAIR hyperintensities restricted to the white matter on the territory of the left middle cerebral artery (MCA), sparing the cortex and basal ganglia (Figure 2A). There was no enhancement with gadolinium, normal apparent diffusion coefficient (ADC), and no magnetic susceptibility artifacts (Figure 2B). The left internal carotid and MCA were normal on repeat CT-angiography. Three EEGs revealed continuous focal slowing of the affected region, without epileptiform abnormalities. The white matter anomalies increased in intensity but followed the same distribution on brain MRI 2 weeks later (Figure 2C). However, they were now associated with low ADC signal (Figure 2D). The patient’s symptoms stopped progressing a few days after admission. She did not improve and was later transferred to a rehabilitation center.

Figure 2: (A) MRI FLAIR sequence demonstrates white matter hyperintensities in the vascular territory of the left MCA on day 18. (B) MRI ADC sequence shows no diffusion restriction of the white matter anomalies. The bilateral periventricular hypointensities correspond to wrap around artifact. (C) MRI FLAIR sequence reveals an increase of the white matter hyperintensities in the vascular territory of the left MCA on day 30. (D) Low signal on MRI ADC sequence on day 30 corresponds to de novo diffusion restriction.
Brain MRI was repeated 2 months later, with resolution of the low ADC signal, persistence of elevated T2/FLAIR, and new T1 hypointensity of the affected white matter. Spectroscopy identified a decreased NAA/choline ratio and a lactate peak. Lumbar puncture revealed slightly elevated proteins (0.74 g/L), with a normal IgG index and no oligoclonal bands. At 3 months, the patient did not improve (NIHSS 8, modified Rankin score 4).
The subacute reappearance of previously resolved symptoms 7 days after discharge cannot be attributed to the direct effects of ischemia at initial presentation. There was no intercurrent illness to suggest a reactivation syndrome mimicking prior stroke symptoms. In the absence of fluctuation and epileptiform anomalies on EEG, the deterioration is not explained by post-stroke non-convulsive seizures.
From an imaging standpoint, the findings are not suggestive of ischemia, as low ADC signal was observed on the second brain MRI performed 1 month after the stroke, and not on the first brain MRI at day 18. In the presence of such widespread white matter anomalies, the grey matter should be involved if the underlying mechanism was ischemia due to a large vessel occlusion. Sparing of the cortex and the basal ganglia suggests an alternative etiology. In the absence of residual thrombus, progressive perforator failure cannot occur multiple days after recanalization and does not explain the involvement of the juxta-cortical white matter, vascularized by distal cortical branches of the MCA.
The differential diagnosis of white matter anomalies with reduction of ADC also includes genetic conditions (phenylketonuria, Menkes), pontine and extra-pontine myelinolysis, toxic leukoencephalopathies (inhalation of heroin vapor, methotrexate), and demyelinating disorders.Reference Citton, Burlina and Baracchini1 In this case, there was no rapid correction of hyponatremia nor exposure to toxins. The localization of the lesions exclusively on the vascular territory of the left MCA and the reappearance of previously resolved symptoms 7 days following recanalization of the artery suggests a pathophysiological relationship with the initial period of brain ischemia.
This is the fourth reported case of delayed onset white matter lesions following recanalized stroke.Reference Sasaki, Tomura and Okada2, Reference Singu, Inatomi, Yonehara and Ando3 One appeared after intravenous thrombolysis and two after endovascular treatment. The presentation after the ischemic episode and the imaging findings suggest delayed onset post-ischemic leukoencephalopathy, reported after cardiac arrest, drug overdose, and carbon monoxide intoxication. However, the ischemic insult being focal rather than global, the deficits and lesions localize to the left MCA territory. While the thrombus occluded the terminal intracranial carotid artery, we hypothesize that the region vascularized by the MCA might have undergone more ischemic stress, possibly due to better collateralization of the anterior cerebral artery.
The delay in the clinical presentation is thought to be due to natural or accelerated depletion of myelin reserves, with symptoms appearing once compromised repair mechanisms are insufficient to compensate for loss of myelin, typically between 7 and 21 days after the initial ischemic insult. Activation of the apoptotic cascade in oligodendrocytes following ischemia could explain the inability to regenerate myelin.Reference Shprecher and Mehta4 A few reports associate the disease with low but not pathological levels of arylsulfatase A (pseudodeficiency).Reference Gottfried, Mayer, Shungu, Chang and Duyn5 However, pseudodeficiency of arylsulfatase A is a common finding in the healthy population (up to 20%) and the association described in the literature does not imply causality.Reference Nelson, Carey and Morris6 The patient’s arylsulfatase A level was normal, but vulnerabilities in other myelin repair pathways could contribute to the pathophysiology of the disease.
With longer delays to recanalization with EVT, the likelihood of delayed apoptosis is theoretically increased as cells undergo longer periods of ischemia before being salvaged. Another hypothetical mechanism is reperfusion injury, but it is unclear why this would occur 1 week after recanalization. In isolation, these explanations are insufficient, as no cases of spontaneously recanalized acute strokes have been reported to follow a similar course, even after prolonged ischemia.
Subacute recrudescence of ischemic symptoms following recanalized stroke is possibly the focal version of post-ischemic leukoencephalopathy. None of the cases published have reported histopathological analyses. Correlating pathology to the already existing descriptions of demyelination in post-ischemic leukoencephalopathy could provide more evidence for the existence of this novel complication following treatment of acute ischemic stroke.
Disclosures
All authors report no disclosures.
Statement of Authorship
AN: study concept and design; drafted the manuscript. A-AP: study concept and design; reviewed the manuscript. FG: study concept and design; reviewed the manuscript. ÉS: study concept and design; reviewed the manuscript.