


Cognitive deficits affecting multiple cognitive domains are a core feature of depressive disorder at all ages. Reference Thomas and O'Brien1 In late-life depression (typically after age 60 years), these deficits have been shown to persist despite recovery from mood symptoms. Reference Bhalla, Butters, Mulsant, Begley, Zmuda and Schoderbek2,Reference Lee, Potter, Wagner, Welsh-Bohmer and Steffens3 Further, structural brain changes in late-life depression have been found to include volume reductions in the medial temporal lobe/hippocampus, Reference Steffens, Byrum, McQuoid, Greenberg, Payne and Blitchington4–Reference Hickie, Naismith, Ward, Turner, Scott and Mitchell6 frontal lobe Reference Schweitzer, Tuckwell, Ames and O'Brien7–Reference Lavretsky, Kurbanyan, Ballmaier, Mintz, Toga and Kumar9 and an increased prevalence of white matter hyperintensities. Reference Herrmann, Le Masurier and Ebmeier10 Several studies have shown that these cerebral changes relate to cognitive deficits in depression: hippocampal volume reductions have been associated with memory deficits Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5,Reference Hickie, Naismith, Ward, Turner, Scott and Mitchell6 and white matter hyperintensities have been associated with impaired memory, Reference Simpson, Jackson, Baldwin and Burns11–Reference Kramer-Ginsberg, Greenwald, Krishnan, Christiansen, Hu and Ashtari13 executive dysfunction Reference Simpson, Jackson, Baldwin and Burns11,Reference Potter, Blackwell, McQuoid, Payne, Steffens and Sahakian14,Reference Sheline, Price, Vaishnavi, Mintun, Barch and Epstein15 and reduced processing speed. Reference Simpson, Jackson, Baldwin and Burns11
However, the relationship between other biological changes in depression such as hypercortisolaemia and brain atrophy and cognitive impairment is less clear. Whereas excessive adrenal glucocorticoid secretion in rodent brains has been shown to lead to hippocampal cell death by excessive glutamate inflow, Reference Sapolsky, Krey and McEwen16 studies in humans have been inconclusive. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5 In addition, few studies have examined which biological and cerebral changes have prognostic utility for the long-term persistence of cognitive impairments in late-life depression. We have previously shown that baseline measures of hippocampal volume predicted persistence of memory impairment after 6 months, whereas levels of cortisol did not. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5 However, the role of white matter hyperintensities was not examined in that study, neither were relations with other cognitive domains.
The aim of the present study was to extend these initial findings to a longer observation period and to examine whether other biological changes are associated with continuing cognitive impairments. We hypothesised that the worse cognitive functioning seen in older adults with depression relative to controls would be associated with more severe white matter hyperintensities and frontal and hippocampal volume reductions at baseline. Based on our previous findings, Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5 we expected hypercortisolaemia to play no role in the persistence of these deficits in the long term.
Method
Participants
Sixty-six people aged 60 and over with DSM–IV 17 major depression were recruited from clinical old age psychiatry services covering geographically based catchment areas, and included referrals from day hospitals, in-patient units and out-patient clinics (depression group). The control group (n = 36) of older people (all over 60 years of age) with no current or past history of depression were recruited from community sources such as The Royal British Legion and spouses of patients attending the same hospital units. After complete description of the study to the participants, written informed consent was obtained. We excluded older adults with a history of prior cognitive impairment, history or evidence of stroke or transient ischemic attack or a Cambridge Cognitive Examination (CAMCOG) Reference Roth, Huppert, Mountjoy and Tym18 score of <70. Participants with severe or unstable physical illness (e.g. insulin-dependent diabetes mellitus, untreated hypothyroidism, uncontrolled heart failure, cancer) were excluded if their conditions were thought to affect their ability to comply with the protocol or have a significant impact on survival that would interfere with completion of the follow-up study. Additional exclusion criteria were: history or current substance/alcohol misuse; long-term use (>2 months) of steroids during lifetime; use of steroid or other medication within the past 3 months thought to interfere with hypothalamic–pituitary–adrenal (HPA) axis; electroconvulsive therapy in past 3 months; use of medication thought to affect cognition (e.g. non-hypnotic benzodiazepines, antipsychotics, sedative tricyclic antidepressants or anticholinergic medication); and presence of other neurological diagnosis. Use of newer antidepressants (e.g. selective serotonin reuptake inhibitors and venlafaxine) and lithium was permitted, as were orally inhaled steroids. The study was approved by the local ethics committee and after a complete description of the study all participants gave written informed consent.
Assessment
Participants underwent a comprehensive assessment including psychiatric history, mental state and physical examination. Depression was diagnosed according to DSM–IV 17 criteria and symptom severity rated with the Montgomery–Åsberg Depression Rating Scale (MADRS). Reference Montgomery and Åsberg19 Demographic information (including past and current medical and psychiatric history, medication taken, family history and education) and history of depressive episodes were collected from multiple sources to validate or enrich information from face-to-face interviews (e.g. case notes, general practitioner records and informant accounts to determine number of previous episodes, age at onset and total lifetime duration of depression). An extensive neuropsychological test battery was administered to consenting participants (detailed below). Participants were reassessed after 6 and 18 months; 93 (90%) and 78 (76%) participants were available for the extended examinations at 6 and 18 months respectively.
Neuropsychological assessment
The test battery was primarily designed to measure memory, processing speed and executive functions, as they represent core neuropsychological deficits in depression in older adults. Reference Thomas and O'Brien1 Tests used in the present study included both traditional pen and paper and computerised tasks.
-
(a) The Rey Auditory Verbal Learning Test (AVLT), Reference Rey20 a test of episodic memory. The three measures immediate recall, delayed recall and delayed recognition (number of correct positives) were used.
-
(b) The FAS verbal fluency test, Reference Lezak, Howieson and Loring21 a task sensitive to frontal lobe impairment.
-
(c) The Trail Making Test (TMT), Reference Lezak, Howieson and Loring21 a test of mental flexibility and divided attention.
-
(d) The Stroop Colour Word Test (SCWT), Reference Stroop22 a test for response inhibition and selective attention.
-
(e) A computerised continuous performance task (VIGIL). Reference Cegalis and Bowlin23 Over 8 min, participants have to press a button to a complex target stimulus (letter K when preceded by the letter A), presented 100 times within a total of 480 stimuli (displayed serially in a pseudo-random fashion). Errors of omission and commission can be used as a measure of vigilance and inhibition but in the present study only response latencies (in msec) were used as a measure of processing speed.
Salivary cortisol analysis
Cortisol levels were assessed by collecting saliva using Salivettes (Sarstedt, Nümbrecht, Germany). Participants chew on a cotton wool plug to produce saliva. Samples were collected at baseline at four time points (08.00 h, 12.00 h, 16.00 h, 20.00 h) over 3 consecutive days, followed by centrifugation and storage at −20°C until assayed. Salivary cortisol was measured using an I125 disequilibrium assay, the radioactive cortisol for which was supplied by Amersham Health, Amersham, UK, the primary antibody Ab1002 and the solid phase anti-rabbit serum by IDS, Tyne and Wear, UK. The intra- and inter-assay coefficients of variation for 7.0, 47 and 87 nmol/l cortisol samples were 13.0 and 14.6%, 10.7 and 9.8%, 9.4 and 10.2% respectively. Average area under the curve (AUC) for the 3 days was calculated. Baseline cortisol data were available for 34 (94%) people in the control group and 42 (64%) participants in the depression group.
Magnetic resonance imaging (MRI) protocol and analysis
As described previously Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5,Reference Lloyd, Ferrier, Barber, Gholkar, Young and O'Brien24 images were acquired using a 1.0 T Siemens Magnetom Impact Expert System (Siemens Medical, Erlangen, Germany). Whole brain T 1-weighted three-dimensional magnetisation prepared rapid-acquisition gradient echo (3D-MPRAGE) turbo flash data-sets were acquired in the sagittal plane (repetition time (TR) = 11.4 msec, echo time (TE) = 4.4 msec, inversion time (TI) = 400 msec, flip angle 15°, matrix 256 × 256, slice thickness 1 mm, cubic voxels of 1 mm) resulting in truly isotropic voxels of 1 × 1 × 1 mm for optimal grey–white matter contrast. Axial dual spin echo sequences were also obtained to yield proton density and T 2 images. Proton density and T 2 images were yielded from RARE (rapid acquisition with relaxation enhancement) technique dual echo (TR = 2800 ms, TE = 14 (proton density) ms/85(T 2) ms, matrix 256 × 256, field of view 230 mm, pixel size 0.92 × 0.92 mm, acquisition time 4 min 13 sec) sequences with axial slice thickness 5 mm and 0.5 mm gap. For volumetric analyses, T 1 images were then transferred to a Sun Ultra 10 work station running Solaris 2.7 (Sun Microsystems, Mountain View, California, USA). Hippocampal volume was analysed by a single operator (A.L.) who was masked to diagnosis using the AnalyzeAVW-3.0 software (AnalyzeDirect. com, Lenexa, Kansas, USA; Mayo Foundation Biomedical Imaging Resource, Rochester, Minnesota, USA). To ensure consistent slicing images were re-orientated along the long axis of the hippocampus (for regional demarcation see O'Brien et al), Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5 whole brain volume was analysed by a semi-automated iterative process of erosion and region-growing. Hippocampal volume was normalised by dividing it by whole brain volume and multiplied by 1000 for convenience of presentation. Whole-brain normalised frontal lobe volume (factor 1000) was analysed with the Medical Information Display and Analysis System (MIDAS–3.0) and analysed by a single operator (O.P.A.) as described previously (Almeida et al). Reference Almeida, Burton, Ferrier, McKeith and O'Brien8 White matter hyperintensities were rated visually from the axial dual echo scans by two experienced raters (R.B. and J.T.O.) using the well-established Scheltens scale. Reference Scheltens, Barkhof, Leys, Pruvo, Nauta and Vermersch25 This can provide a measure of overall severity of white matter hyperintensities by summing scores for the four regions covered by the scale (periventricular, deep white matter, basal ganglia and infratentorial) on the rating scale. Severity in white matter hyperintensities could be successfully rated in 27 (75%) people in the control group and 51 (76%) in the depression group. Magnetic resonance images of brain volumes could be successfully analysed in 35 (97%) controls and 51 (76%) individuals with depression.
Statistical analysis
Neuropsychological test scores were standardised in relation to the controls. An overall memory z-score was created by adding up the AVLT z-scores of immediate recall, delayed recall and delayed recognition, and this ‘compound memory score’ was again standardised. Similarly, an overall executive functions z-score was created by adding up the z-scores of verbal fluency, TMT difference A–B and SCWT correct responses. As a result, we produced three summary measures of cognitive function, with higher scores indicating better performance: memory, executive functions and processing speed (VIGIL latencies). We analysed the impact of our biological measures on the difference in cognitive outcome between the control group and the depression group by testing the interaction between group (0, control group; 1, depression group) and the biological measures on the neuropsychological z-scores. Analyses were adjusted a priori for age, gender and years of education. The alpha-level was fixed at P<0.05 and all statistical tests reported are two-tailed. In addition, we report P-values derived from Simes' modification of the Bonferroni correction in order to control for multiple testing. Reference Simes26 Simes' procedure is less conservative than traditional Bonferroni correction and is therefore considered superior if outcomes are correlated with each other, as in this study. A test is considered significant if the observed P is lower than Simes' adjusted significance level. All analyses were performed on STATA 10.1 for Windows.
Results
At 18 months, 45 out of 66 (68%) people in the depression group were available for assessment, of which none met DSM–IV criteria for dementia. Of those lost to follow-up (Fig. 1), 19 withdrew consent and 3 died. There were no differences between participants available and not available for follow-up in relation to age (t = 0.10, d.f. = 65, P = 0.919), gender (χ2= 0.07, d.f. = 65, P = 0.797), years of education (t = −0.17, d.f. = 65, P = 0.785), age at onset (t = −0.04, d.f. = 65, P = 0.972), MADRS score (baseline: t = −1.47, d.f. = 65, P = 0.147; 6 months: t = 0.34, d.f. = 55, P = 0.739) or remission status (baseline: χ2 = 0.53, d.f. = 65, P = 0.466; 6 months: χ2 = 1.04, d.f. = 55, P = 0.308). Similarly, there were no differences between individuals in the depression group who were or were not available for follow-up on z-scores for memory (baseline: t = 1.12, d.f. = 32, P = 0.270; 6 months: t = 1.11, d.f. = 49, P = 0.274), executive functions (baseline: t = 1.19, d.f. = 35, P = 0.241; 6 months: t = 1.87, d.f. = 49, P = 0.067) or processing speed (baseline: t = 0.83, d.f. = 28, P = 0.414; 6 months: t = 0.72, d.f. = 44, P = 0.478). Finally, there were no differences between these two groups on cortisol concentration (t = 0.82, d.f. = 40, P = 0.415), whole brain volume (t = −1.78, d.f. = 49, P = 0.082), hippocampal volume (t = 0.10, d.f. = 49, P = 0.921), frontal lobe volume (t = −0.07, d.f. = 49, P = 0.942) or overall white matter hyperintensities (t = −0.67, d.f. = 49, P = 0.508).
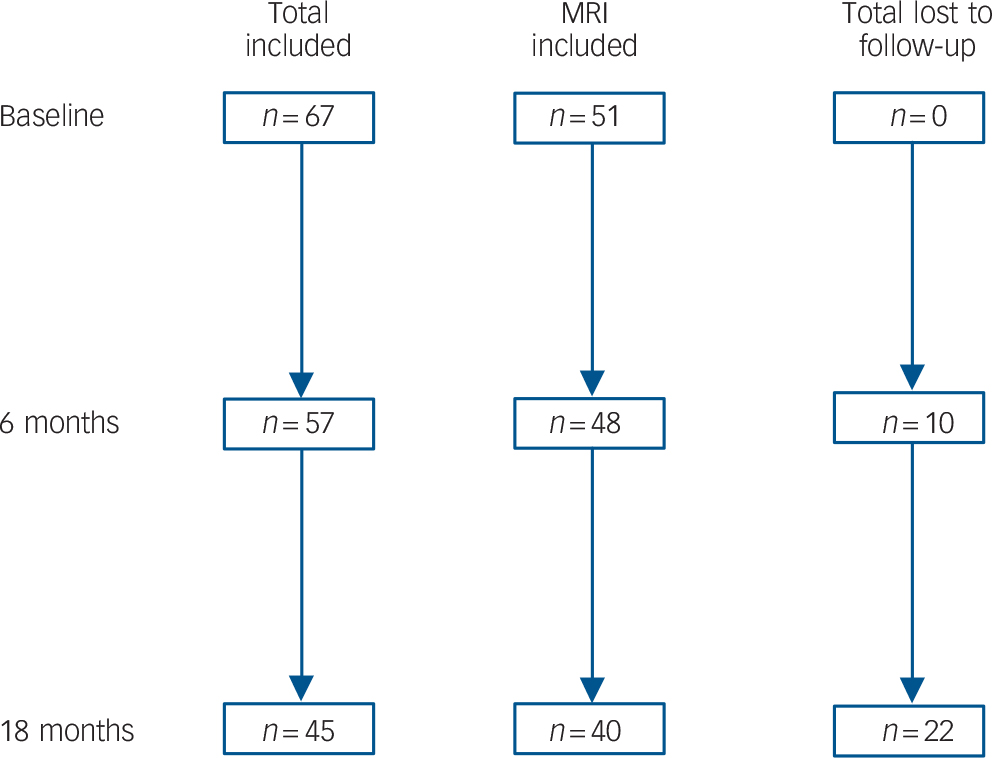
Fig. 1 Flow chart illustrating loss to follow-up and numbers of participants retained in the total sample and those who had baseline magnetic resonance imaging (MRI).
Baseline MRI data and follow-up neuropsychological test scores were available for 35 people in the depression group and 29 people in the control group. Individuals in the depression group on average had fewer years of formal education and higher depression scores at baseline and follow-up compared with those in the control group (Table 1). They performed worse on all neuropsychological tests at follow-up and had higher AUC cortisol levels at baseline, but there were no significant differences in total, frontal or hippocampal brain volume and overall white matter hyperintensities.
Table 1 Differences in demographic and clinical characteristics between the healthy control group and the depression group who had data available from baseline magentic resonance imaiging or cortisol and follow-up neuropsychological testing

| Depression group (n = 35) | Healthy control group (n = 29) | Analysis | |||
|---|---|---|---|---|---|
| Test statistic | d.f. | P | |||
| Age, years: mean (s.d.) | 74.1 (6.5) | 72.8 (6.9) | t = -0.81 | 62 | 0.422 |
| Female: n (%) | 28 (80) | 22 (76) | χ2 = 0.16 | 62 | 0.690 |
| Education, years: mean (s.d.) | 9.4 (2.0) | 10.6 (2.2) | t = 2.14 | 62 | 0.037 |
| Montgomery—Åsberg Depression Rating Scale at baseline, mean (s.d.) | 20.6 (9.8) | 2.0 (2.1) | t = -10.01 | 62 | ≤ 0.001 |
| Montgomery—Åsberg Depression Rating Scale at 18 months, mean (s.d.) | 8.0 (10.2) | 1.6 (2.0) | t = -3.32 | 62 | 0.002 |
| Illness duration, weeks: mean (s.d.) | 58.3 (56.0) | ||||
| Number of episodes, mean (s.d.) | 3.6 (3.2) | ||||
| Antidepressant use, n (%) | 28 (80) | ||||
| Memory at 18 months, mean (s.d.) | -1.7 (2.1) | 0.1 (0.9) | t = 4.19 | 62 | ≤ 0.001 |
| Executive functions at 18 months, mean (s.d.) | -1.3 (1.8) | 0.0 (1.0) | t = 3.26 | 60 | 0.002 |
| Processing speed at 18 months, mean (s.d.) | -0.7 (1.2) | 0.1 (1.0) | t = -2.71 | 58 | 0.009 |
| Cortisol at baseline, mean (s.d.) | 158 (80) | 100 (24) | t = -3.63 | 43 | ≤ 0.001 |
| Whole brain volume (in mm3), mean (s.d.) | 965 (81) | 969 (88) | t = 0.20 | 61 | 0.844 |
| Left + right raw frontal volume (in mm3), mean (s.d.) | 96 (16) | 98 (14) | t = 0.52 | 62 | 0.609 |
| Left + right normalised frontal volume,a mean (s.d.) | 92 (12) | 92 (7) | t = 0.07 | 62 | 0.944 |
| Left + right raw hippocampal volume (in mm3), mean (s.d.) | 5.6 (0.9) | 5.8 (0.8) | t = 0.55 | 61 | 0.583 |
| Light + right normalised hippocampal volume,b mean (s.d.) | 5.8 (0.7) | 5.9 (0.5) | t = 0.63 | 61 | 0.533 |
| Overall white matter hyperintensities, mean (s.d.) | 12.2 (5.4) | 13.2 (9.9) | t = 0.47 | 53 | 0.642 |
Impact of biological measures on group differences in neurocognitive function
Tests for interactions between group and biological measures on neurocognitive outcome adjusted for age, gender and years of education are summarised in Table 2. Differences in memory z-scores at 18 months between the depression group and the control group were not moderated by cortisol levels or volumetric measures, but showed a significant interaction with overall white matter hyperintensities. With every one-point increase in white matter hyperintensities severity, the score on memory declined by 0.21 standard deviations in individuals with depression. Likewise, there were no significant interactions between group and cortisol levels or volumetric measures on executive function z-scores, but yet again there was a significant interaction with overall white matter hyperintensities (decline of 0.18 standard deviations with every one-point increase in white matter hyperintensities severity). Both effects remained significant when multiple testing was controlled for. In contrast, group differences in processing speed were not moderated by any biological measure.
Table 2 Interactions between baseline biological measures and group (0, healthy control group; 1, depression group) on neurocognitive outcomes at 18 months

| z-scores | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Memory | Executive function | Processing speed | |||||||
| Interaction by group | 95% CI | P | Interaction by group | 95% CI | P | Interaction by group | 95% CI | P | |
| Cortisol | 0.01 | -5.17 to 1.12 | 0.505 | -0.01 | -0.04 to 0.01 | 0.288 | 0.01 | -0.01 to 0.02 | 0.393 |
| Whole brain volume | 0.00 | -0.01 to 0.01 | 0.593 | 0.00 | -0.01 to 0.01 | 0.709 | 0.00 | -0.00 to 0.01 | 0.575 |
| Frontal lobe volume | -0.03 | -0.14 to 0.09 | 0.636 | -0.05 | -0.15 to 0.04 | 0.279 | -0.02 | -0.10 to 0.05 | 0.554 |
| Hippocampal volume | 0.47 | -1.00 to 1.95 | 0.521 | 0.94 | -0.36 to 2.24 | 0.153 | 0.16 | -0.82 to 1.14 | 0.741 |
| Overall white matter hyperintensities | -0.21 | -0.32 to -0.10 | ≤ 0.001a | -0.18 | -0.29 to -0.06 | 0.004b | -0.03 | -0.12 to 0.05 | 0.460 |
Further explorations into the role of white matter hyperintensities
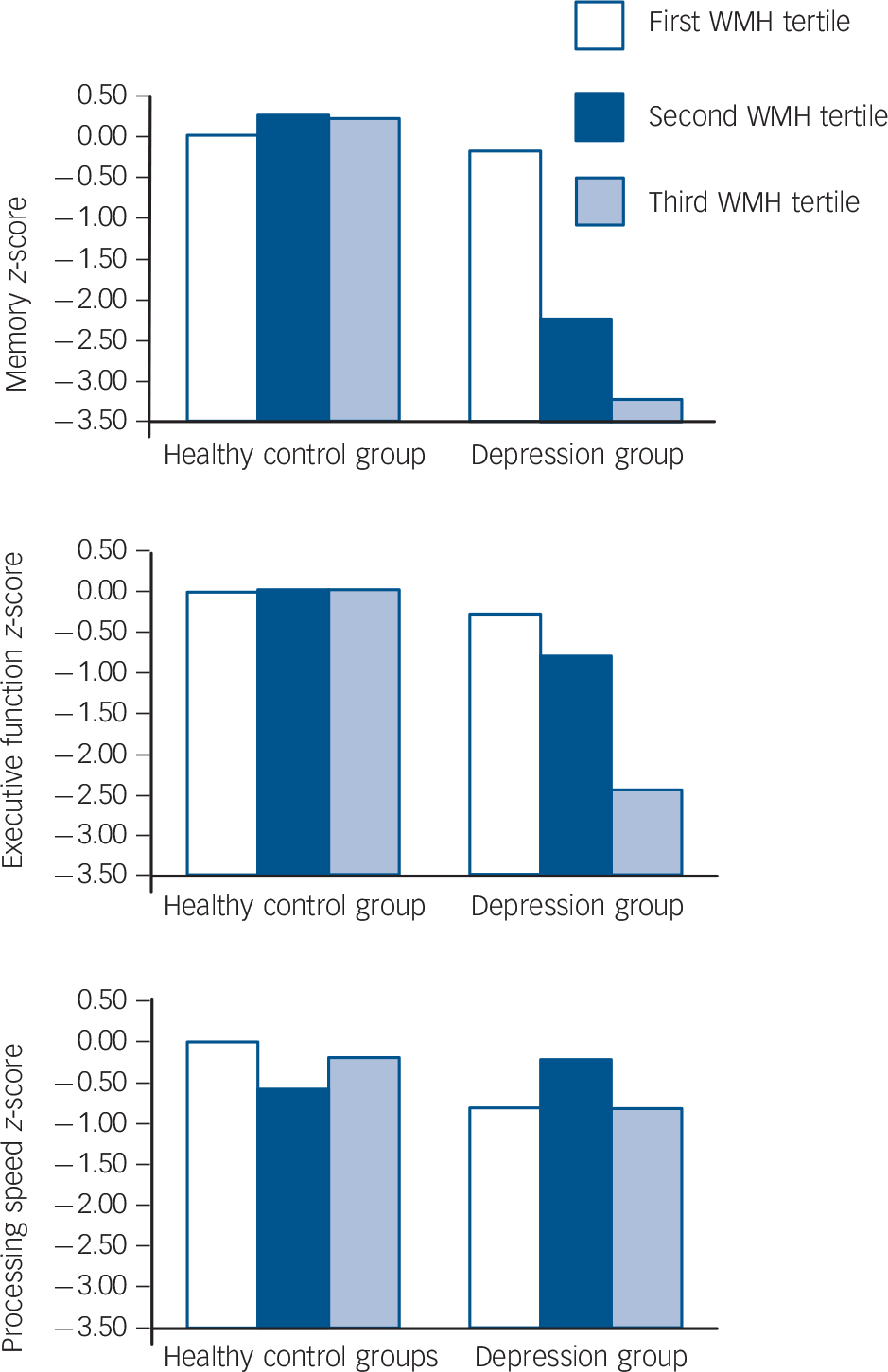
In order to clarify the role of white matter hyperintensities further, we investigated potential dose–response relations between white matter hyperintensities severity and neurocognitive outcome by dividing the overall severity rating into tertiles. There were significant interactions between group and white matter hyperintensities tertiles on memory and executive function scores that showed evidence of an effect gradient (Fig. 2). Using the healthy controls within the first tertile (no/minor white matter hyperintensities, n = 10) as the reference group, individuals with depression within the same tertile (n = 11) did not differ in memory scores (z-score difference with reference group: −0.18, 95% CI −1.41 to 1.06, P = 0.775). However, being depressed within the second white matter hyperintensities tertile (n = 11) was associated with a decline of two standard deviations in memory (z-score difference with reference group: 2.21, 95% CI −3.68 to −0.74, P = 0.004; test for interaction of group and white matter hyperintensities tertile (first v. second): b = −2.04, 95% CI −3.92 to −0.14, P = 0.36). Being depressed in the third white matter hyperintensities tertile (n = 11) went together with a decline of three standard deviations (z-score difference −3.23, 95% CI = −4.56 to −1.91, P=0.001; test for interaction of group and white matter hyperintensities tertile (first v. third): b = −3.06, 95% CI −4.84 to −1.27, P=0.001). For executive functions, neither the participants with depression within the first white matter hyperintensities tertile group (z-score difference −0.28, 95% CI −1.45 to 0.91, P = 0.643) nor those within the second white matter hyperintensities tertile differed from the reference group (z-score difference −0.77, 95% CI −2.18 to 0.64, P = 0.276; test for interaction: b = −0.50, 95% CI −2.31 to 1.31, P = 0.583). Only white matter hyperintensities within the third tertile were associated with worse executive functions scores in participants with depression (z-score difference −2.40, 95% CI −3.72 to −1.08, P≤0.001; test for interaction: b = −2.12, 95% CI −3.86 to −0.39, P = 0.018).

Fig. 2 Differences in neurocognition across groups and across tertiles of white matter hyperintensities (WMH).
In order to clarify the role of the anatomical location of white matter hyperintensities, the continuous overall severity rating was divided into its constituent parts: deep white matter hyperintensities, periventricular white matter hyperintensities, basal ganglia hyperintensities and infratentorial hyperintensities. Adjusted analyses showed that group difference in memory z-scores were moderated by deep (test for interaction: b = −0.30, 95% CI −0.47 to −0.13, P≤0.001) and periventricular white matter hyperintensities (test for interaction: b = −0.68, 95% CI −1.21 to −0.15, P = 0.014), but not basal ganglia (test for interaction: b = −0.07, 95% CI −0.50 to 0.35, P = 0.729) or infratentorial hyperintensities (test for interaction: b = −0.32, 95% CI −1.07 to 0.43, P = 0.398). Group differences in executive functions were moderated by deep white matter hyperintensities (test for interaction: b = −0.21, 95% CI = −0.40 to −0.02, P = 0.027), but not periventricular white matter hyperintensities (test for interaction: b = −0.46, 95% CI −0.99 to 0.06, P = 0.082), basal ganglia (test for interaction: b = −0.07, 95% CI −0.46 to 0.32, P = 0.708) or infratentorial hyperintensities (test for interaction: b = −0.39, 95% CI −1.07 to 0.30, P = 0.260).
Discussion
Main findings
In the present study, we found robust associations between ratings of white matter hyperintensities and neurocognitive impairment among older people with depression who were followed up for 18 months. More severe white matter lesions, especially in the deep white matter and periventricular regions, were associated with greater deficits in memory and executive functions at follow-up compared with a healthy control group. In contrast, whole or regional brain volume, including the hippocampus, and baseline cortisol levels were unrelated to persisting cognitive deficits. Further, white matter hyperintensities predicted cognitive deficits in people with depression but were unrelated to cognitive performance in healthy controls. This confirms earlier reports Reference Sheline, Price, Vaishnavi, Mintun, Barch and Epstein15 and is in line with studies showing that white matter lesions are commonly present in the brains of healthy older adults, where they do not necessarily relate to poor cognitive outcome. Reference Fernando and Ince27 Several mechanisms might explain this. First, individuals with depression might have lowered brain reserve to compensate for the damage exerted by white matter hyperintensities as a result of the effects of accumulated genetic and environmental risk exposure. As a consequence, the additional burden placed by white matter hyperintensities gives rise to cognitive deficits in this group. Alternatively, the neuropathology of white matter hyperintensities might be different in depression compared with normal ageing. Indeed, previous work has shown that white matter hyperintensities in depression are more often ischaemic in nature. Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria28 Third, the strategic anatomical lesion location, and consequently the neurocircuits disrupted, might differ between the groups. Late-life depression is particularly associated with deep white matter lesions affecting projection to and from the frontal lobes, thereby disrupting circuits for cognitive and emotional control, leading to depressive symptoms and executive dysfunction. Reference Sheline, Price, Vaishnavi, Mintun, Barch and Epstein15
Our findings are consistent with previous cross-sectional reports showing total brain hyperintensities correlate with impaired episodic memory Reference Simpson, Jackson, Baldwin and Burns11–Reference Kramer-Ginsberg, Greenwald, Krishnan, Christiansen, Hu and Ashtari13 and executive functions in older people with depression. Reference Simpson, Jackson, Baldwin and Burns11,Reference Potter, Blackwell, McQuoid, Payne, Steffens and Sahakian14,Reference Sheline, Price, Vaishnavi, Mintun, Barch and Epstein15 In a recent report we found patients' deficits in individual cognitive domains to be stable from baseline to 6 and 18 months, Reference Köhler, Thomas, Barnett and O'Brien29 thus suggesting that white matter hyperintensities contribute to the persistence of cognitive dysfunction observed among some older people with depression. These findings are consistent with the vascular hypothesis of depression, Reference Alexopoulos30 and indicate that white matter hyperintensities and cerebrovascular disease play an important role in the phenotypic expression of depression in later life. Reference Herrmann, Le Masurier and Ebmeier10,Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria28 The aetiological relevance of vascular factors in the onset or recurrence of depression has been implicated by research showing that white matter hyperintensities can pre-date the onset of depression, Reference Teodorczuk, O'Brien, Firbank, Pantoni, Poggesi and Erkinjuntti31,Reference Godin, Dufouil, Maillard, Delcroix, Mazoyer and Crivello32 and are likely to further progress over time in people with prevalent depression. Reference Godin, Dufouil, Maillard, Delcroix, Mazoyer and Crivello32 In addition, more severe white matter changes have been associated with worse prognosis of depression, including poor response to antidepressant treatment, Reference Alexopoulos, Murphy, Gunning-Dixon, Latoussakis, Kanellopoulos and Klimstra33,Reference Hickie, Scott, Mitchell, Wilhelm, Austin and Bennett34 chronic and relapsing course, Reference O'Brien, Ames, Chiu, Schweitzer, Desmond and Tress35 an increased risk for dementia and higher mortality. Reference Baldwin, Walker, Simpson, Jackson and Burns36 These findings have led to the consideration of ‘vascular depression’ as a distinct diagnostic subtype in the upcoming DSM–V associated with old age, white matter hyperintensities, late onset and deficits in executive functions. Reference Sneed, Roose and Sackeim37 Recently, Sneed and colleagues, using latent class analysis, reported that the presence of deep white matter lesions has almost perfect discriminating ability for distinguishing classes of vascular and non-vascular depression. Reference Sneed, Rindskopf, Steffens, Krishnan and Roose38 Our study adds that such white matter hyperintensities have predictive validity as they are associated over 18 months with poorer memory and executive functions. However, the predictive validity of white matter hyperintensities for treatment response, clinical and other cognitive outcomes has to be clarified further before any criteria of vascular depression can be recommended for use in regular clinical practice.
The present study provides important evidence that although white matter hyperintensities are associated with key cognitive outcomes, other biological abnormalities, including hypercortisolaemia, are not. Despite evidence for a toxic effect of chronic glucocorticoid hypersecretion on hippocampal neurons in animal models Reference Sapolsky, Krey and McEwen16 and humans, Reference Lupien, de Leon, de Santi, Convit, Tarshish and Nair39 we found no effect of baseline cortisol levels or hippocampal volume on differences between participants with depression and healthy controls in follow-up neurocognition. As we reported previously, cortisol levels are further not associated with hippocampal volume or baseline memory function. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier5 In another report using this same sample, we found that participants with a late onset but not an early onset had reduced hippocampal volume, Reference Lloyd, Ferrier, Barber, Gholkar, Young and O'Brien24 which has been replicated by other volumetric studies Reference Hickie, Naismith, Ward, Turner, Scott and Mitchell6,Reference Ballmaier, Narr, Toga, Elderkin-Thompson, Thompson and Hamilton40 and contradicts the idea that cumulative (lifetime) cortisol exposure is sufficient to explain hippocampal damage in depression. We acknowledge that our results are not consistent with some reports in younger people with depression Reference Sheline, Sanghavi, Mintun and Gado41 and thus suggest that in older people with depression pathogenic mechanisms other than hypercortisolaemia might explain hippocampal volume loss and associated memory deficits, including vascular pathology.
Strengths and weaknesses
Notable strengths of this study are a comprehensive assessment of biological parameters that have been suggested to have an impact on cognitive functioning in depression, a relatively long follow-up duration, the assessment of core cognitive domains, and the inclusion of a matched-comparison group of healthy participants that was followed-up in parallel. Yet, some methodological aspects deserve attention. Magnetic resonance image analyses of different regions of interest were performed using different software packages, although correlations of whole brain volumes obtained using both packages was very high (r = 0.91). Ratings of white matter hyperintensities were based on visual rather than volumetric data, although this approach has been shown to be reliable and to have good face validity. Reference Lloyd, Ferrier, Barber, Gholkar, Young and O'Brien24 Salivary cortisol is a measure with high variability, so that the absence of an effect on cognition might relate to levels not accurately reflecting average HPA axis function in a participant. However, we attempted to reduce that variability by measuring cortisol at four time points over 3 days at each time point, and used the average AUC to gain a measure of overall cortisol output. In addition, we did not find an association between hippocampal volume and cognition, which further undermines the relevance of cortisol-induced brain changes. It would have further been interesting to look at the relevance of white matter hyperintensities on cognition as a function of age at onset since vascular changes have been found to relate to late-onset depression in particular. Reference Herrmann, Le Masurier and Ebmeier10 Unfortunately, we were unable to explore this further given the small cell sizes that would have resulted from stratification by age at onset. However, there were no significant differences in the number of early-onset or late-onset participants across white matter hyperintensities tertiles in a post hoc analysis (highest white matter hyperintensities tertile: early-onset depression, 4 (22%); late-onset depression, 7 (47%); Fisher's exact P = 0.292).
Implications
In conclusion, white matter hyperintensities moderate the severity of continuing cognitive deficits in older people with depression in a dose–response fashion. In contrast, high levels of cortisol and hippocampal volume loss do not appear to play a major role in the persistence of depression-related cognitive deficits. Our results suggest a need to find effective treatment strategies for these deficits which remain following successful treatment of mood symptoms.
Acknowledgements
We thank Philip English for expert radiographer support, and Nicky Barnett and Liz McGuckin for help with participant recruitment and assessment.




eLetters
No eLetters have been published for this article.